Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 3
Theoretical Study on the Corrosion Inhibitive Effect of Phenyl and Alkynyl Derivatives of Benzo[a]phenothiazin-5-one and Benzo[a]phenoxazin-5-one Dyes
Efeturi A Onoabedje1, Francis K Ojo2* and Kovo G Akpomie1
1Department of Pure & Industrial Chemistry, University of Nigeria, Nsukka, Nigeria
2Department of Chemical Sciences, Bingham University, Karu, Nigeria
- *Corresponding Author:
- Francis K Ojo
Department of Chemical Sciences
Bingham University, Karu, Nigeria
Abstract
The theoretical inhibitive effect of 6-phenyl-5H-benzo[a]phenothiazin-5-one, 6-phenyl-5H-benzo[a]phenoxazin-5-one and 6-(2-phenylethynyl)- 5H-benzo[a]phenoxazin-5-one compounds on mild steel in hydrochloric acid solution was described. Compounds 6-phenyl-5H-benzo[ a]phenothiazin-5-one and 6-phenyl-5H-benzo[a]phenoxazin-5-one were obtained as brilliantly reddish and yellowish powdered solids via palladium catalyzed cross-coupling of phenyl boronic acid with 6-chloro-5H-benzo[a]phenothiazin-5-one, 6-chloro-5H-benzo[a]phenoxazin-5- one respectively. The reaction of ethynylbenzene with 6-chloro-5H-benzo[a]phenoxazin-5-one afforded orange coloured solid. The intense colors of these dyes and their ease of re-oxidation of Na2S2O4-reduced derivatives make them suitable as vat dyes. In addition, quantum chemical calculations based on DFT/B3LYP level confirm that the substitution of the oxygen in the hetero cyclic ring with sulphur and the bonding of an alkynyl group (triple bond) in conjugation to the aromatic ring facilitates the adsorption of the organic compound toward the metal surface with the highest inhibition efficiency obtained for 6-(2-phenylethynyl)-5H-benzo[a]phenoxazin-5-one.
Keywords
Phenoxazine, Phenothiazine, Dyes, Corrosion, Theoretical
Introduction
The control of corrosion can be achieved by several methods. One of the most effective alternatives for the protection of metallic surfaces against corrosion is by the use of inhibitors [1]. Inorganic compounds such as phosphates, nitrites, hydrazine and chromates adopted proved to be harmful to the environment, thus nesseciating the need for the search of green inhibitors [2]. A perusal of the literature on corrosion inhibitors reveals that most organic inhibitors employed as corrosion inhibitors contain nitrogen, oxygen, sulphur and/or aromatic ring in their molecular structure [3-6]. Also the theoretical prediction of the efficiency of corrosion inhibitors in selecting excellent corrosion inhibitors have continuously gained popularity especially with the significant progress in the building of sophisticated software packages which has being employed in quantum mechanical calculations [7]. This work is of paramount importance because the organic inhibitors are affordable, nontoxic, easy to synthesize and inhibitors have not being reported as corrosion inhibitors. The aim of this paper is to extend these investigations in order to discuss the relationship between quantum chemical calculations inhibition efficiencies of the inhibitors by determining the quantum chemical parameters such as the Energies of Highest Occupied Molecular Orbital (EHOMO) and the Lowest Unoccupied Molecular Orbital (ELUMO), the energy difference (ΔE) between EHOMO and ELUMO, Log P, polarizability, molecular volume and molecular weight.
Materials and Methods
Chemical synthesis of inhibitors
The 6-phenyl-5H-benzo[a]phenothiazin-5-one, 6-phenyl-5H-benzo[a]phenoxazin-5-one and 6-(2-phenylethynyl)-5H-benzo[a]phenoxazin-5-one compounds used in this study were synthesised as described in our previous works [8-10].
Reduction of compounds 3-5
In a typical experiment compound 3 (323 mg, 1 mmol) was placed in a reaction flask containing water (2 ml) and acetone (40 ml). Sodium dithionite (500 mg) was added and the mixture was refluxed in a water bath for 1.5 h. During the refluxing period the colour changed from red to yellow. The entire mixture was poured into a solution containing 0.5 g of dithionite in 100 ml of ice cold water and stirred. This was quickly filtered by suction but before the product could be collected from the filter paper it had turned reddish. Analysis of the product confirmed it to be the starting iminoquinoid compound 6. This shows the reduced compound was auto-oxidized under this condition. Similar observations were seen in the case of compounds 4 and 5.
Theoretical method
All the quantum chemical calculations were performed with SPARTAN’10. The full optimization was initially achieved by using Molecular Mechanics Force Fields (MMFF). The results from MMFF were further selected as input and re-optimized using Semi empirical AM1. The Semi empirical AM1 structures were selected as input and were re-optimized using Density Functional Theory (DFT) at the level of B3LYP methods which uses the exchange functional proposal by Becke and all the correlations functional given by Lee, Yang and Parr. The 6-31G* basis set has been used in conjunction with DFT method because it has the advantage of being flexible enough to guarantees reliable theoretical results and being small enough for rapid calculations. It represents an excellent compromise between completeness and economy. The molecular geometry was fully optimized without any constraint using analytical gradient procedure implemented within the program package [7-10]. The following quantum chemical indices were considered: the energy of the EHOMO, ELUMO, separation energy (ELUMO–EHOMO), Log P and polarizability. The chemical structures and labelled optimized geometry of these organic inhibitors are shown in Figures 1 and 2 respectively.
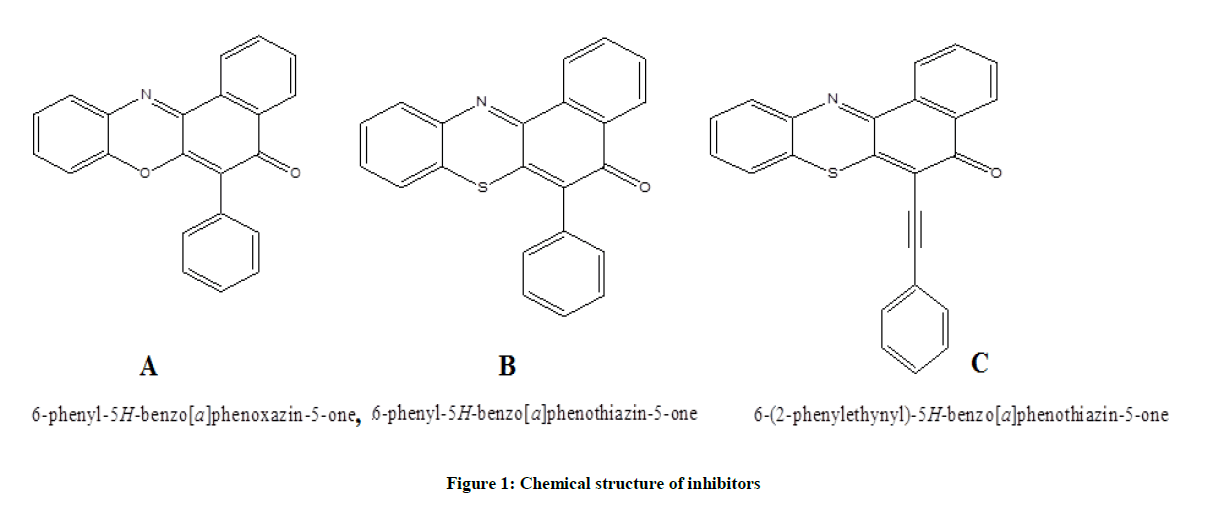
Figure 1: Chemical structure of inhibitors
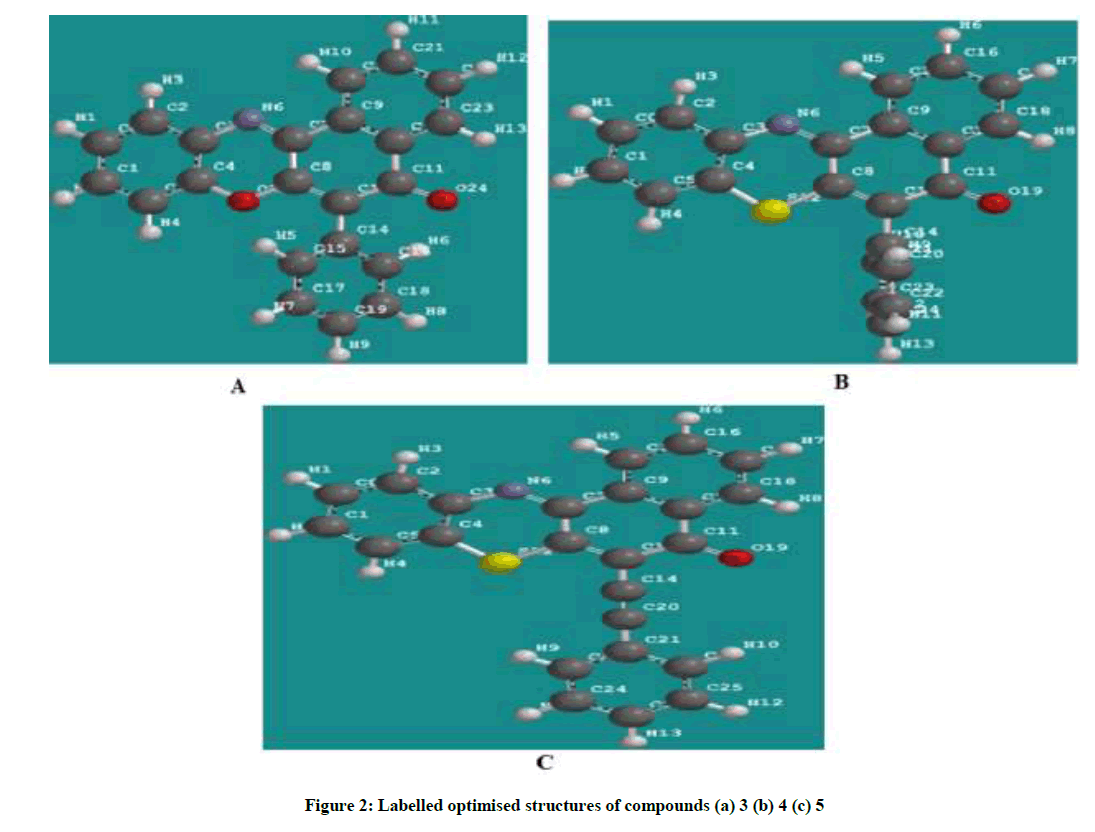
Figure 2: Labelled optimised structures of compounds (a) 3 (b) 4 (c) 5
Result and Discussion
The organic compounds 1-5 used in this study were synthesised as described in our previous work [8-10] (Scheme 1). The coupling of 2- aminophenol and 2-aminothiophenol with 2,3-dichlroro-1,4-napthiquinone under provided compounds 1 and 2.
The 6-phenyl-5H-benzo[a]phenoxazin-5-one 3 and 6-phenyl-5H-benzo[a]phenothiazin-5-one 4 were obtained as brilliantly reddish and yellowish powdered solids via palladium catalyzed cross-coupling of phenyl boronic acid with 6-chloro-5H-benzo[a]phenoxazin-5-one 1 and 6- chloro-5H-benzo[a]phenothiazin-5-one 2 respectively. The reaction of ethynylbenzene with compound 1 afforded orange coloured solid identified as 2-(6-ethynylbenzene-5H-benzo[a]phenothiazin-5-one 5. The spectral and analytical data were consistent with the assigned structures. Figure 3 showed the 1H & 13C-NMR of compounds 3-5.
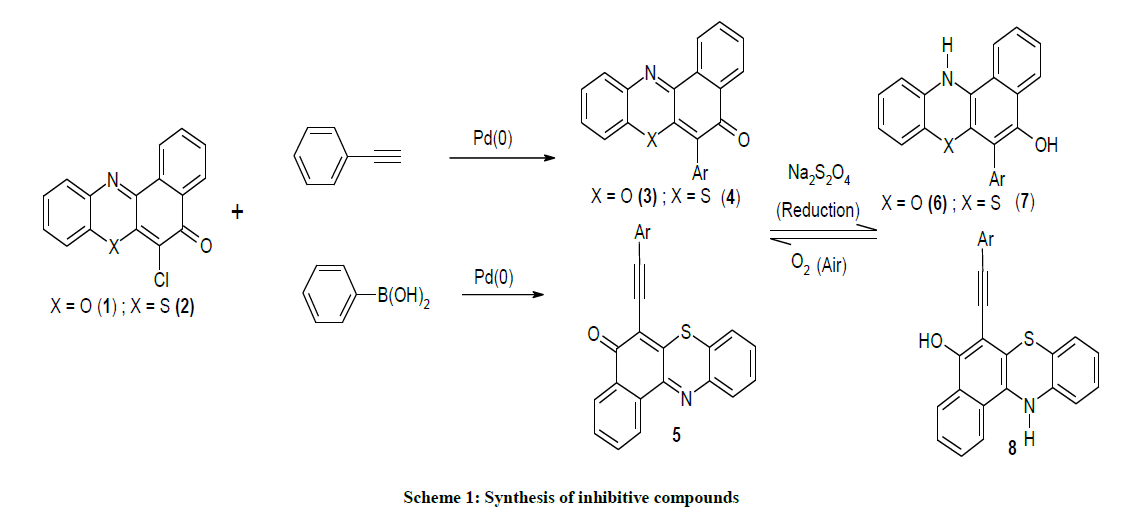
Scheme 1: Synthesis of inhibitive compounds
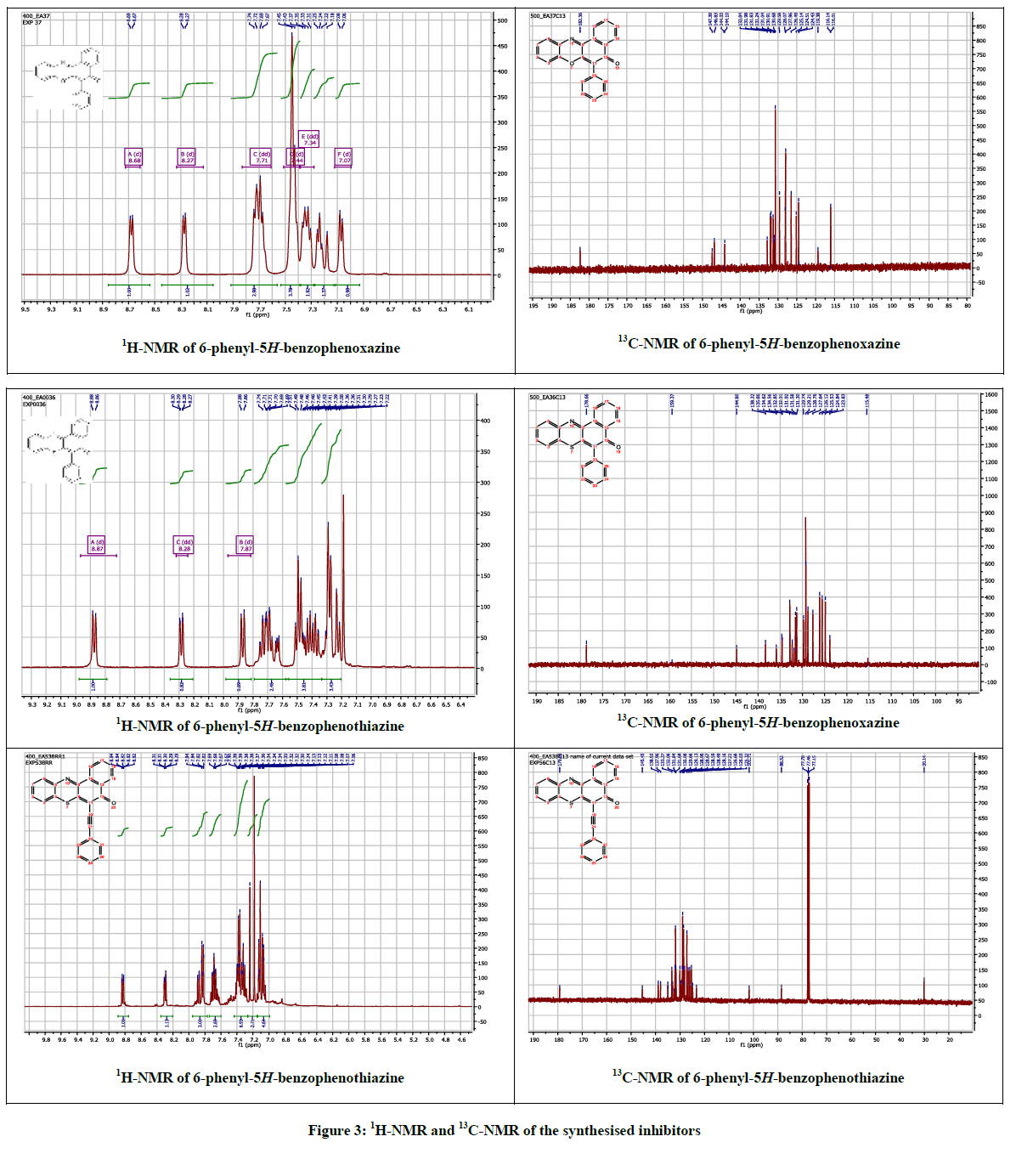
Figure 3: 1H-NMR and 13C-NMR of the synthesised inhibitors
The reduction of compounds 3, 4 and 5 with sodium dithionite afforded unstable benzo[a]phenothiazin-5-ol 4 and benzo[a]phenoxazine-5-ols which were not isolated in pure form as they rapidly reverted to the original compounds in air indicating their possible application as vat dyes [11-14]. Quantum chemical parameters obtained from the calculations which are responsible for the inhibition efficiency of inhibitors such as the energies of frontier molecular orbitals (EHOMO and ELUMO), separation energy (ELUMO–EHOMO), the net charge on the amino group, substituent constant (log P), polarizability, molecular volumes and molecular weights are tabulated in Table 1.
| Quantum chemical parameters | 3 | 4 | 5 |
|---|---|---|---|
| EHomo (eV) | -5.71 | -5.44 | -5.45 |
| ELumo (eV) | -2.77 | -2.85 | -2.87 |
| ELumo–EHomo (eV) | 2.94 | 2.59 | 2.58 |
| log P | 3.55 | 3.82 | 4.39 |
| Polarizability | 67.35 | 69.8 | 70.55 |
| Volume (Ǻ3) | 328.74 | 357.92 | 367.18 |
| Weight (amu) | 323.35 | 347.37 | 363.44 |
Table 1: Quantum chemical parameters of organic inhibitors derivatives using DFT method
The EHOMO is often associated with the electron donating ability of molecule [15-17]. Therefore, increasing values of EHOMO indicated higher tendency for donation of electrons to the appropriate acceptor molecule with low energy and empty molecular orbital, thus increasing values of EHOMO facilitate the adsorption of the inhibitor [16]. The ELUMO indicates the ability of the molecule to accept electrons. Hence, the lower the value of ELUMO the more apparent for the molecule to accept electrons [18]. The separation energy which indicates reactivity is the energy difference between the HOMO and the LUMO. The energy gap also relates to how soft or hard a molecule is. A larger energy gap indicates low reactivity to chemical specie, thus a soft molecule is more reactive than a hard molecule [16,19]. The binding ability of the inhibitor to the metal surface increases with increasing the energy of HOMO, decreasing energy of LUMO and low separating energy values. Polarizability is the ratio of induced dipole moment to the intensity of the electric field. The induced dipole moment is proportional to polarizability [20]. log P (Substituent constant) are empirical quantities which account for the variation of the structure and do not depend on the parent structure but vary with the substituent. Hence increasing the value of log p increases the inhibition efficiencies of organic inhibitors [21].
Molecular volume and weight are quantum parameters that determine molecular size and effective surface coverage. These invariably determine how effective a molecule can be adsorbed on and cover metal surface, thereby isolating it from the corroding environment. As the value of these parameters increase, so also the likely corrosion inhibition potentials of the molecules increase [14].
The calculated quantum chemical parameters showed that cpd 3 has a lower HOMO energy, higher LUMO energy and higher separation energy as compared to cpd 4 and 5. This could be attributed the presence of the sulphur atom cpd 4 and additional presence of triple bond in cpd 5. This effect of sulphur atom and additional triple bond makes available more non-bonding pair electrons and π electrons which facilitate the adsorption of the inhibitor onto the mild steel surface through donating and back donating processes. The calculated substituent constant (log P) and polarizability show a gradual increase with the highest values being that of inhibitor 5, Table 1. Inhibitor 5 has the highest molecular volume, 367.18 Ǻ3 and molecular weight of 363.440 amu due to the sulphur analogue and additional alkenyl group (triple bond) which probably makes a better surface coverage of the metal surface which explains the highest inhibition efficiency for the inhibitor 3.
Figures 4 and 5 showed the HOMO and LUMO orbital plots for the studied molecules at the B3LYP/6-31G level. The electron rich region is red and the electron poor region is blue in the molecules. As usual, the red regions identify the most negative potentials and the most electron-rich regions. It is observed that the total electron density surfaces for the studied organic molecules reveals that the highest levels of electron density are found around the hetero cyclic ring, double bond oxygen and the triple bond carbon. In summary the above results indicate that the organic inhibitors are adsorbed on the metal surface through the hetero cyclic ring, double bond oxygen and the triple bond carbon.
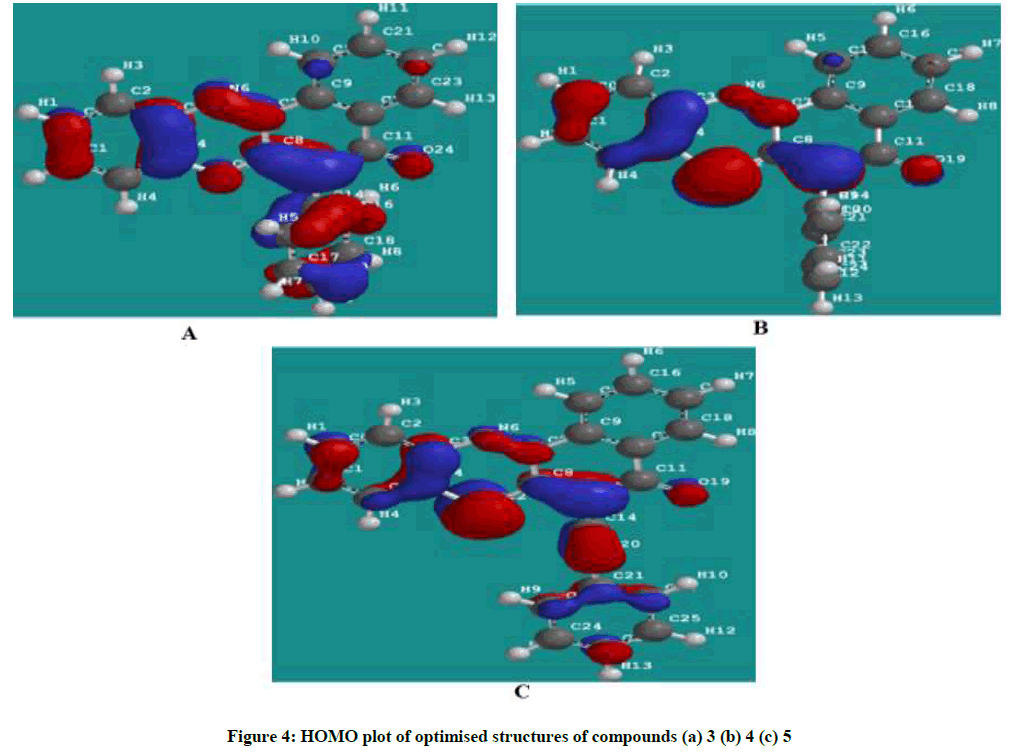
Figure 4: HOMO plot of optimised structures of compounds (a) 3 (b) 4 (c) 5
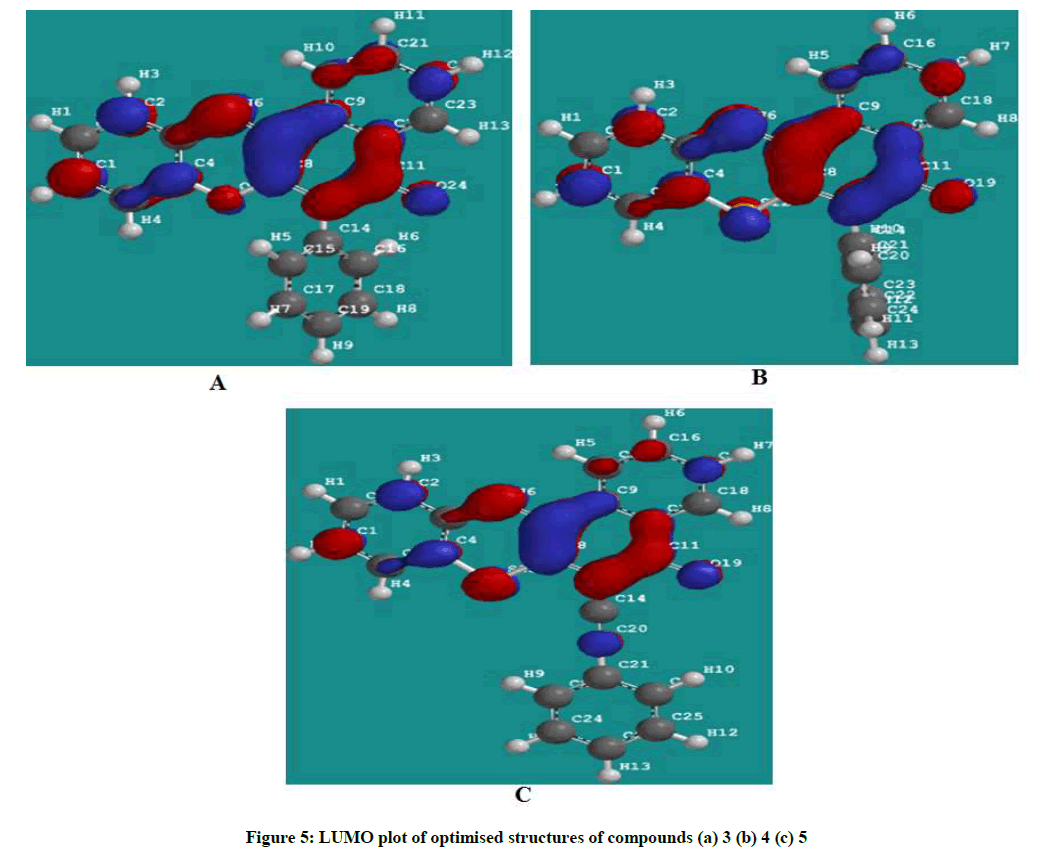
Figure 5: LUMO plot of optimised structures of compounds (a) 3 (b) 4 (c) 5
Acknowledgement
We thank Petroleum Technology Development Funds (PTDF) for financial support and Cardiff University for a short-term research opportunity granted to Dr. E.A. Onoabedje.
Conclusion
Quantum chemical calculations based on DFT/B3LYP level were performed to find the relation between the molecular structure of the inhibitor and the inhibition efficiency. The calculated quantum chemical indices confirm that the substitution of the oxygen in the hetero cyclic ring with sulphur and the bonding of an alkynyl group (triple bond) in conjugation to the aromatic ring facilitates the adsorption of the organic compound toward the metal surface with the highest inhibition efficiency obtained for molecule 3.
References
- F. Matjaz, J. Jennifer, Corrosion Science.,2014,86, 17-41.
- L.A. Nnanna, I.O. Owate, E.E. Oguzie, Int. J. Mater. Eng., 2014, 4(5), 171-179.
- R. Hasanov, M. Sadıkog ˘lu, S. Bilgiç, Appl. Surf. Sci., 2007, 253, 3913-3919.
- M. Özcan, R. Solmaz, G. Kardas, I. Dehri, Coll. Surf. A: Physicochem. Eng. Aspects.,2008, 325, 57-67.
- K. Bhrara, H. Kim, G. Singh, Corros. Sci., 2008, 50, 2747-2753.
- M. Behpour, S.M. Ghoreishi, N. Soltani, M. Salavati-Niasari, M. Hamadanian, A. Gandomi, Corros. Sci., 2008, 50, 2172-2180.
- G. Gokhan, Corros. Sci., 2008, 50, 2981-2992.
- E.A. Onoabedje, U.C. Okoro, A. Sarkar, D.W. Knight, J. Heterocyc. Chem., 2016, 53(6), 1787-1794.
- E.A. Onoabedje, U.C. Okoro, A. Sarkar, D.W. Knight, J. Sulfur Chem., 2016, 34(3), 269-281.
- E.A. Onoabedje, U.C. Okoro, D.W. Knight, J. Heterocyc. Chem., 2017, 54(1), 206-214.
- C.O. Okafor, U.C. Okoro, Dyes Pigm., 1988, 9, 427-442.
- G. Gokhan, B. Semra, Corros. Sci.,2009, 51, 1876-1878.
- I.B. Obot, N.O. Obi-Egbedi, S.A. Umoren, Int. J. Electrochem. Sci.,2009, 4, 863-877.
- R.M. Issa, M.K. Award, F.M. Atlam, Mater. Corros., 2009, 60, 813-819.
- S. Xia, M. Qiu, L. Yu, F. Liu, H. Zhao, Corros. Sci., 2008, 50, 2021-2029.
- E.E. Ebenso, D.A. Isabirye, N.O. Eddy, Int. J. Mol. Sci.,2010, 11, 2473-2498.
- T. Arslan, F. Kandemirli, E.E. Ebenso, I. Love, H. Alemu, Corros. Sci.,2009, 51, 35-47.
- E.E. Ebenso, T. Arslan, F. Kandemirli, I. Love, C. Ogretir, S. Murat, S.A. Umoren, Int. J. Quantum Chem., 2010, 110, 2614-2636.
- E.E. Ebenso, T. Arslan, F. Kandemirli, N. Caner, I. Love, Int. J. Quantum Chem., 2010, 110, 1003-1018.
- C.T. Wang, S.H. Chen, H.Y. Ma, C.S. Qi, J. Appl. Electrochem., 2003, 33, 179-186.
- G. Gece, Corros. Sci., 2011, 53, 3873-3898.



