Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 8
The Investigation of Spectral and Theoretical Properties of 2-(3-Cyclopropyl-4,5-dihydro-1H-1,2,4-triazol-5-on-4-yl-iminomethyl) Benzoic Acid by Using B3LYP/HF 6-31g (d,p) Basis Set
Medetalibeyoğlu H*, Özdemir G and Yüksek H
Department of Chemistry, Kafkas University, 36100 Kars, Turkey
- *Corresponding Author:
- Medetalibeyoğlu H
Department of Chemistry
Kafkas University
36100 Kars, Turkey
Abstract
1H-1,2,4-triazole has been theoretically studied. All theoretical calculations have been carried out by using Density Functional Theory (DFT) and Hartree-Fock (HF) methods. The vibrational frequencies were calculated by using HF/6-31G(d,p) and DFT(B3LYP)/6-31G(d,p) basis sets in ground state. The UV-vis (ethanol), Proton Nuclear Magnetic Resonance (1H-NMR) and Carbon-13 Nuclear Magnetic Resonance (13C-NMR) spectra of title molecule have been recorded. The 1H and 13C-NMR chemical shifts of the title molecule were calculated by the GIAO method and compared with experimental results. Using gauge independent atomic orbital method 1H and 13C-NMR chemical shifts have been calculated and correlated with the experimental chemical shifts. The polarizability (α), hyperpolarizability (β), dipole moment along with molecular electrostatic potential surface have been calculated. The molecular electrostatic potential (MEP) map was calculated to assign reactive site on the surface of the molecule. The calculated electronic, structural (bond lengths and bond angles) and several thermodynamic parameters, molecular electrostatic potential (MEP) map of the compound were performed using the Hartree-Fock (HF) and density functional method (DFT/B3LYP) with 6-31G(d,p) basis set.
Keywords
1H-1,2,4-triazol-5-one, GIAO, DFT/HF, 6-31G(d,p) basis set
Introduction
4-Amino-1H-1,2,4-triazole and their derivatives are reported to possess a wide spectrum of biological activities such as antifungal, antibacterial, anticancer, anti-inflammatory and antitumor properties [1-13]. During the past two decades, considerable attention has been paid to the chemistry and computational chemical calculations of 1,2,4-triazole and their derivatives. That’s why these methods have been commonly used the prediction of many properties in the chemical systems. The computational chemical models are widely used design of functional materials. Also, many authors have studied the structure, spectroscopic, electronic and thermodynamic parameters of many organic compounds by using theoretical calculation methods [13-18].
The derivatives of 1,2,4-triazole are known to exhibit many biological activities. Hence, various 1,2,4-triazole and their derivatives have been theoretically analyzed [18-21]. Reliable results consistent with experimental results have been obtained for 1,2,4-triazole and their derivatives [21-25]. For this purpose, firstly, all quantum chemical calculations of target compound have been carried out by using B3LYP/6-31G(d,p) and HF/6-31G(d,p) methods. The DFT/B3LYP and HF methods are known to be the most widely used methods in many reported references [26,27]. Also, the vibrational frequencies and calculated structural parameters (bond lengths and bond angles), electronic, thermodynamic parameters, gauge including atomic orbital (GIAO) 1H and 13C chemical shift values, UV-vis, HOMO-LUMO energies, charge distributions, total static dipole moment (μ), the mean polizability (<α>), the anisotropy of the polarizability (Δα), the mean first-order hyperpolarizability (<β>), electronegativity(χ), hardness(η), molecular electrostatic potential maps (MEP) of target molecule were calculated by using the optimized structures with B3LYP/6-31G(d,p) and HF/6-31G(d,p) basis sets in ground state. The vibrational frequencies of the title molecule were related with the spectral data obtained with DFT/B3LYP and HF 6-31G(d,p) basis sets. In the identification of calculated IR data was used the veda4f program [28]. The calculated vibrational frequencies were compared with their experimental data.
Material and Methods
Theoretical
The quantum chemical calculations of target molecule were performed by using Gaussian 09W [29] and GaussView [30] software. The molecular geometry of title compound was optimized using density functional theory (DFT/B3LYP) and hartree fock (HF) methods with the 6-31G(d,p) basis set in ground state. From the optimized geometry, structural (bond lengths and bond angles), 1H and 13C-NMR chemical shift values, UV-Vis values, total energy, molecular electrostatic potential (MEP) map, dipole moment and Mulliken atomic charges, vibrational frequencies and HOMO–LUMO energies of the molecule were calculated with B3LYP/ HF 6-31G(d,p) levels. The IR, Proton Nuclear Magnetic Resonance (1H-NMR), Carbon-13 Nuclear Magnetic Resonance (13C-NMR) and UV-vis (ethanol) spectra of target molecule have been recorded. The veda4f program was used for the identification of the calculated IR data [28]. The gauge independent atomic orbital (GIAO) 1H and 13C-NMR chemical shift values was calculated at B3LYP/6-31G(d,p) and HF/6-31G(d,p) levels [31,32]. The theoretical UV-vis spectral values were carried out using TD-DFT method in ethanol solvent. The experimental spectra values of this compound obtained from 1H and 13C-NMR spectra were compared to the calculated results from the DFT/B3LYP and HF 6-31G(d,p) basis sets.
Optimized geometries
2-(3-Cyclopropyl-4,5-dihydro-1H-1,2,4-triazol-5-on-4-yl-iminomethyl) benzoic acid was optimized by means of DFT(B3LYP)/HF methods with 6-31G(d,p) basis set. The optimized geometric structure of 2-(3-cyclopropyl-4,5-dihydro-1H-1,2,4-triazol-5-on-4-yl-iminomethyl) benzoic acid is shown in Figure 1. The geometric parameters (bond angels and bond lengths), Mulliken atomic charges of 2-(3-cyclopropyl-4,5-dihydro-1H-1,2,4-triazol-5-on-4-yl-iminomethyl) benzoic acid were calculated using the DFT(B3LYP)/HF 6-31G(d,p) method. The calculated parameters are given in Tables 1-3 and Chart 1.
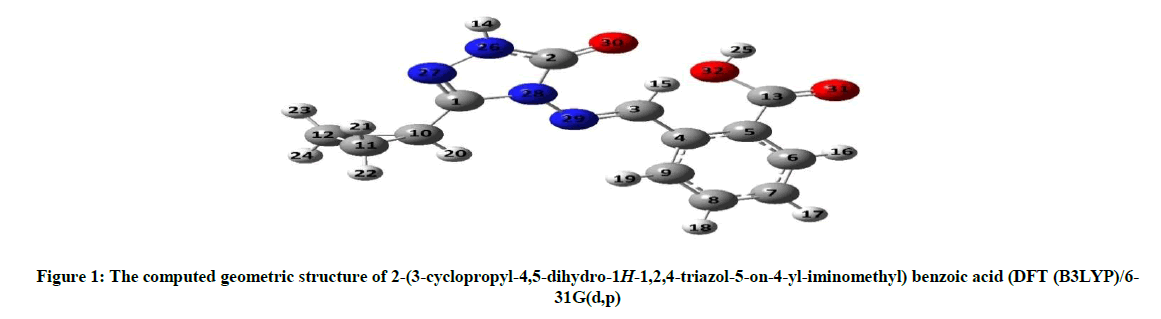
Figure 1: The computed geometric structure of 2-(3-cyclopropyl-4,5-dihydro-1H-1,2,4-triazol-5-on-4-yl-iminomethyl) benzoic acid (DFT (B3LYP)/6-31G(d,p)
| S. No. | Bond Angels (0) | B3LYP 6-31G(d,p) | HF 6-31G(d,p) |
|---|---|---|---|
| 1 | C(1)-N(27)-N(26) | 104.633 | 105 |
| 2 | C(1)-N(28)-N(29) | 121.645 | 121.404 |
| 3 | C(1)-N(28)-C(2) | 108.268 | 108.133 |
| 4 | C(1)-C(10)-H(20) | 113.423 | 113.89 |
| 5 | C(1)-C(10)-C(11) | 119.225 | 119.18 |
| 6 | C(1)-C(10)-C(12) | 119.217 | 108.291 |
| 7 | H(20)-C(10)-C(11) | 118.069 | 118.209 |
| 8 | C(10)-C(11)-H(21) | 116.357 | 117.218 |
| 9 | C(10)-C(11)-H(22) | 117.21 | 116.905 |
| 10 | H(21)-C(11)-H(22) | 115.188 | 114.952 |
| 11 | H(21)-C(11)-C(12) | 117.677 | 117.938 |
| 12 | H(22)-C(11)-C(12) | 118.913 | 118.487 |
| 13 | C(11)-C(12)-C(10) | 60.578 | 60.65 |
| 14 | C(11)-C(12)-H(23) | 117.683 | 117.745 |
| 15 | C(11)-C(12)-H(24) | 118.99 | 118.984 |
| 16 | H(23)-C(12)-H(24) | 115.2 | 114.868 |
| 17 | H(23)-C(12)-C(10) | 116.383 | 117.231 |
| 18 | H(24)-C(12)-C(10) | 117.216 | 117.183 |
| 19 | C(12)-C(10)-H(20) | 117.578 | 116.731 |
| 20 | N(27)-C(1)-N(28) | 111.284 | 111.155 |
| 21 | N(27)-N(26)-H(14) | 120.438 | 120.939 |
| 22 | N(26)-C(2)-O(30) | 129.935 | 129.398 |
| 23 | H(14)-N(26)-C(2) | 124.993 | 125.2 |
| 24 | O(30)-C(2)-N(28) | 128.814 | 128.625 |
| 25 | C(2)-N(28)-N(29) | 130.053 | 130.392 |
| 26 | N(28)-N(29)-C(3) | 118.64 | 119.431 |
| 27 | N(29)-C(3)-H(15) | 122.026 | 122.026 |
| 28 | N(29)-C(3)-C(4) | 118.482 | 118.482 |
| 29 | H(15)-C(3)-C(4) | 119.489 | 119.489 |
| 30 | C(3)-C(4)-C(5) | 122.991 | 122.991 |
| 31 | C(4)-C(5)-C(13) | 126.052 | 126.052 |
| 32 | C(3)-C(4)-C(9) | 118.891 | 118.891 |
| 33 | C(5)-C(13)-O(31) | 123.874 | 123.874 |
| 34 | C(5)-C(13)-O(32) | 114.44 | 114.44 |
| 35 | O(31)-C(13)-O(32) | 121.65 | 121.65 |
| 36 | C(13)-O(32)-H(25) | 106.072 | 106.072 |
| 37 | C(4)-C(5)-C(6) | 119.621 | 119.621 |
| 38 | C(13)-C(5)-C(6) | 114.322 | 114.322 |
| 39 | C(5)-C(6)-H(16) | 117.747 | 117.747 |
| 40 | C(5)-C(6)-C(7) | 121.264 | 121.264 |
| 41 | H(16)-C(6)-C(7) | 120.987 | 120.987 |
| 42 | C(6)-C(7)-H(17) | 120.073 | 120.073 |
| 43 | C(6)-C(7)-C(8) | 119.396 | 119.396 |
| 44 | H(17)-C(7)-C(8) | 120.531 | 120.987 |
| 45 | C(7)-C(8)-H(18) | 120.246 | 120.073 |
| 46 | C(7)-C(8)-C(9) | 120.019 | 119.396 |
| 47 | H(18)-C(8)-C(9) | 119.727 | 119.727 |
| 48 | C(8)-C(9)-C(4) | 121.634 | 121.634 |
| 49 | C(8)-C(9)-H(19) | 120.385 | 120.385 |
| 50 | H(19)-C(9)-C(4) | 117.973 | 117.973 |
Table 1: The calculated bond angles (0) of title compound (6-31G(d,p) B3LYP/HF)
| S. No. | Bond Lengths | B3LYP 6-31G(d,p) | HF 6-31G (d,p) |
|---|---|---|---|
| 1 | C(1)-N(28) | 1.39 | 1.38 |
| 2 | C(1)-N(27) | 1.31 | 1.27 |
| 3 | C(1)-C(10) | 1.47 | 1.48 |
| 4 | C(10)-H(20) | 1.08 | 1.07 |
| 5 | C(10)-C(11) | 1.52 | 1.5 |
| 6 | C(11)-H(21) | 1.09 | 1.07 |
| 7 | C(11)-H(22) | 1.08 | 1.08 |
| 8 | C(11)-C(12) | 1.5 | 1.49 |
| 9 | C(12)-H(23) | 1.09 | 1.08 |
| 10 | C(12)-H(24) | 1.09 | 1.08 |
| 11 | C(12)-C(10) | 1.52 | 1.51 |
| 12 | N(27)-N(26) | 1.38 | 1.37 |
| 13 | N(26)-H(14) | 1.01 | 1 |
| 14 | N(26)-C(2) | 1.37 | 1.35 |
| 15 | C(2)-O(30) | 1.22 | 1.22 |
| 16 | C(2)-N(28) | 1.42 | 1.42 |
| 17 | N(28)-N(29) | 1.37 | 1.37 |
| 18 | N(29)-C(3) | 1.29 | 1.26 |
| 19 | C(3)-H(15) | 1.08 | 1.08 |
| 20 | C(3)-C(4) | 1.48 | 1.49 |
| 21 | C(4)-C(5) | 1.42 | 1.4 |
| 22 | C(5)-C(13) | 1.5 | 1.5 |
| 23 | C(13)-O(31) | 1.22 | 1.19 |
| 24 | C(13)-O(32) | 1.35 | 1.33 |
| 25 | O(32)-H(25) | 0.97 | 0.95 |
| 26 | C(5)-C(6) | 1.4 | 1.39 |
| 27 | C(6)-H(16) | 1.08 | 1.07 |
| 28 | C(6)-C(7) | 1.39 | 1.38 |
| 29 | C(7)-H(17) | 1.09 | 1.07 |
| 30 | C(7)-C(8) | 1.39 | 1.38 |
| 31 | C(8)-H(18) | 1.09 | 1.08 |
| 32 | C(8)-C(9) | 1.39 | 1.38 |
| 33 | C(9)-H(19) | 1.08 | 1.07 |
| 34 | C(9)-C(4) | 1.41 | 1.39 |
Table 2: The calculated bond lengths (A0) of title compound (6-31G(d,p) B3LYP/HF)
| Atoms | DFT | HF | Atoms | DFT | HF |
|---|---|---|---|---|---|
| C1 | 0.59 | 0.65 | H17 | 0.10 | 0.17 |
| C2 | 0.82 | 1.05 | H18 | 0.10 | 0.16 |
| C3 | 0.13 | 0.18 | H19 | 0.11 | 0.18 |
| C4 | -0.05 | -0.04 | H20 | 0.13 | 0.18 |
| C5 | -0.17 | -0.15 | H21 | 0.14 | 0.17 |
| C6 | -0.10 | -0.11 | H22 | 0.12 | 0.14 |
| C7 | -0.09 | -0.15 | H23 | 0.14 | 0.15 |
| C8 | -0.08 | -0.13 | H24 | 0.12 | 0.14 |
| C9 | -0.10 | -0.13 | H25 | 0.33 | 0.37 |
| C10 | -0.14 | -0.20 | N26 | -0.43 | -0.56 |
| C11 | -0.21 | -0.25 | N27 | -0,37 | -0.37 |
| C12 | -0.21 | -0.24 | N28 | -0.45 | -0.65 |
| C13 | 0.53 | 0.79 | N29 | -0.32 | -0.30 |
| H14 | 0.29 | 0.34 | O30 | -0.54 | -0.66 |
| H15 | 0.18 | 0.24 | O31 | -0.47 | -0.56 |
| H16 | 0.13 | 0.20 | O32 | -0.49 | -0.61 |
Table 3: The calculated Mulliken atomic charges of title compound
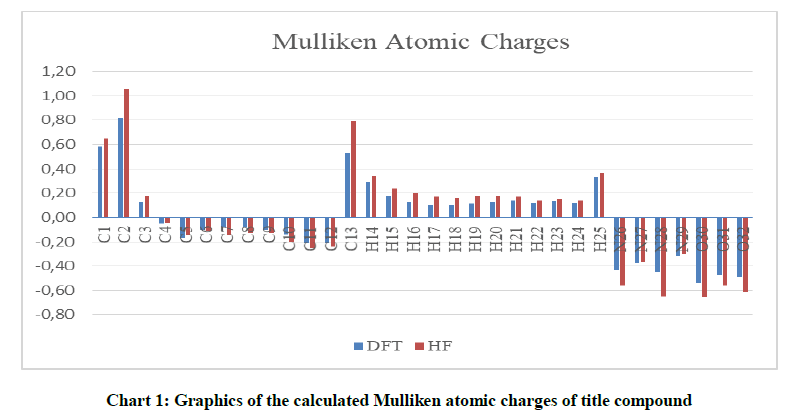
Chart 1: Graphics of the calculated Mulliken atomic charges of title compound
The calculated C4-C5, C5-C6, C6-C7, C7-C8, C8-C9, C9-C4 bond lengths of benzene rings in this compound are [1.42/1.40 A0], [1.40/1.39 A0], [1.39/1.38 A0 ], [1.39/1.39 A0], [1.39/1.38 A0] and [1.41/1.38 A0], respectively. The Ar(C)-Ar(C) bond lengths of benzene rings are generally observed at 1.34-1.53 A0 in literature [33,34]. The calculated C-H bond lengths of the compound are about 1.08 A0 and the C-H bond lengths in literature are 1.09 A0 [33,34]. Also, the calculated N26-C2 bond length in 1,2,4-triazole-5-one ring is [1.37/1.35 A0]. It has been recorded that it has bond length between single bonded N-N and double bonded N=N due to resonance. The same bond length in literature is recorded as between 1.29-1.47 A0 [33,34]. Compared with the bond lengths in the literature, the results show that the molecular structure is very well.
It has been recorded that the electronegative N and O atoms have negative atomic charge values in gas phase. The carbon atoms surrounded with electronegative atoms have positive atomic charge values in gas phase. The C1 atom which is surrounded with two electronegative atoms (N, N), C1 atom surrrounded with three electronegative atoms (N, N, O) have the highest positive charges values. All hydrogen atoms of the compound have positive atomic charge values (Table 3 and Chart 1).
NMR, IR and UV-vis spectra
The GIAO 1H and 13C chemical shift, experimental and calculated IR, UV-vis values were determined by employing DFT(B3LYP)/HF 6-31G(d,p) method. The calculational and experimental results are given in Figures 2 and 3, Tables 4-7 and Chart 2.
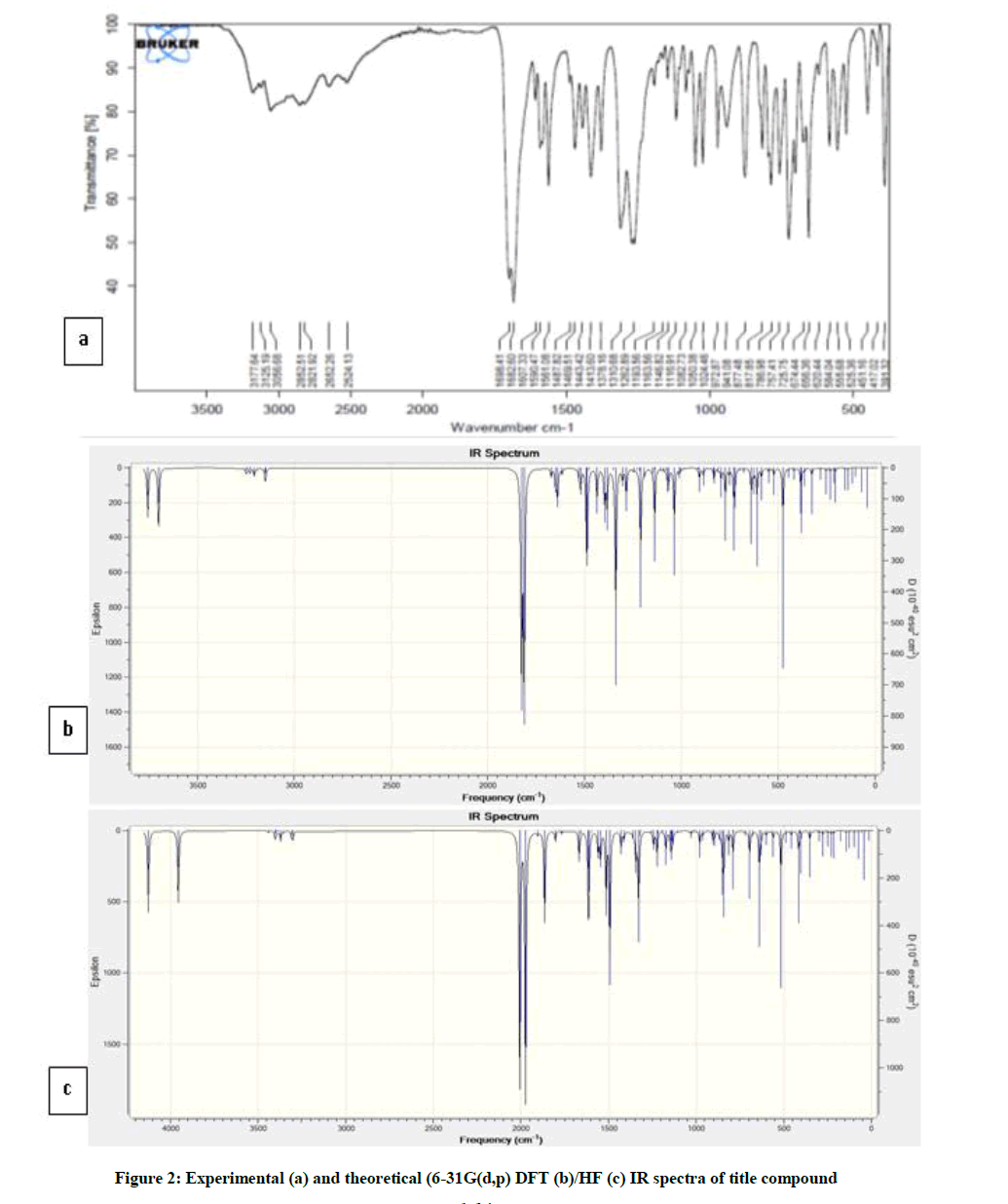
Figure 2: Experimental (a) and theoretical (6-31G(d,p) DFT (b)/HF (c) IR spectra of title compound
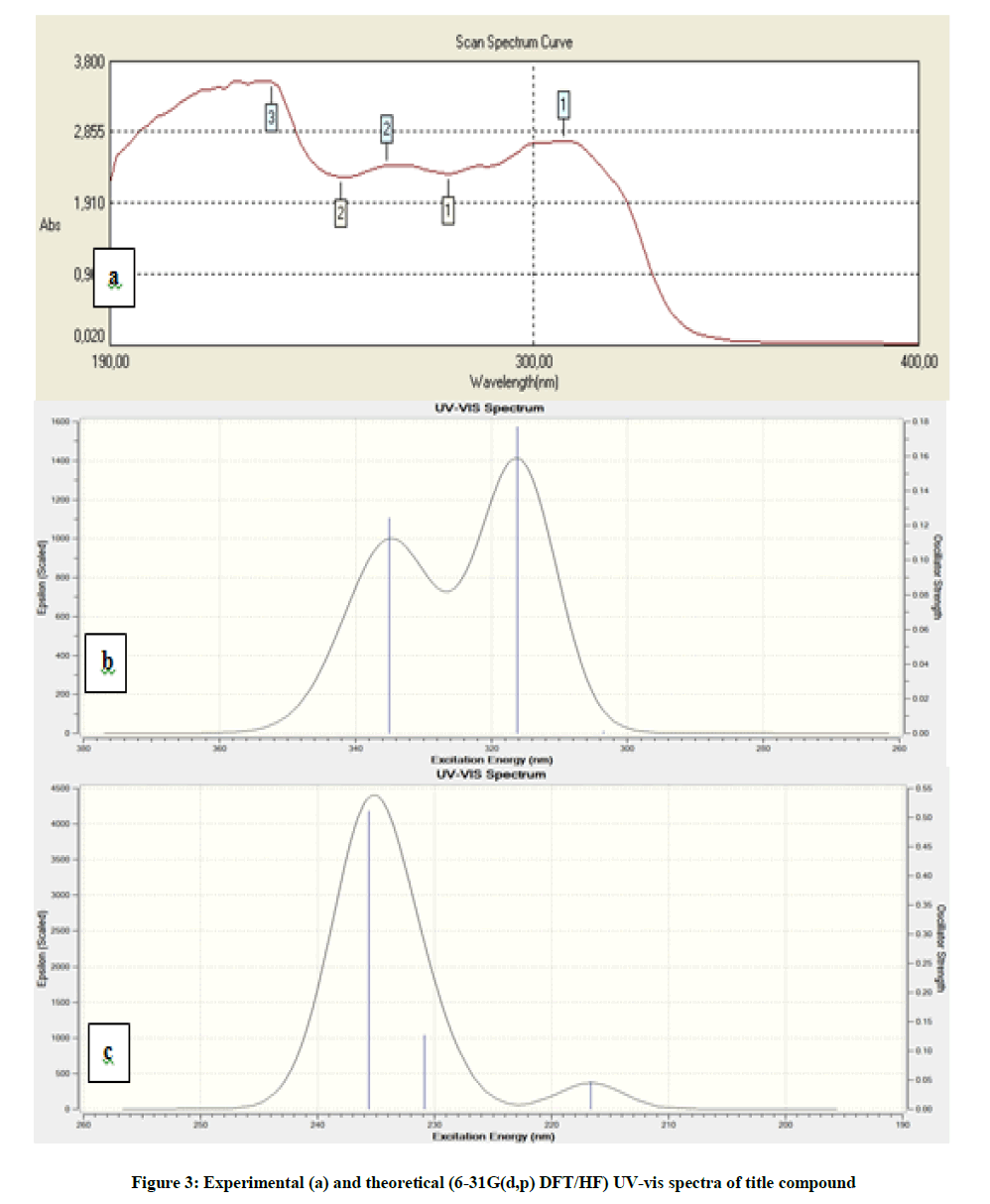
Figure 3: Experimental (a) and theoretical (6-31G(d,p) DFT/HF) UV-vis spectra of title compound
| δExp. | δcal. HF (Vacum) | δcal. HF (DMSO) | Different | Different (DMSO) | δcal. B3LYP (Vacum) | δcal. B3LYP (DMSO) | Different | Different (DMSO) | |
|---|---|---|---|---|---|---|---|---|---|
| C1 | 148.25 | 154.23 | 155.38 | -5.98 | -7.13 | 148.1 | 149.65 | 0.15 | -1.40 |
| C2 | 151.15 | 151.77 | 152.56 | -0.62 | -1.41 | 145.78 | 146.57 | 5.37 | 4.58 |
| C3 | 152.86 | 154.85 | 155.14 | -1.99 | -2.28 | 149.76 | 150.43 | 3.10 | 2.43 |
| C4 | 133.67 | 140.95 | 140.35 | -7.28 | -6.68 | 134.61 | 133.81 | -0.94 | -0.14 |
| C5 | 130.73 | 134.4 | 133.92 | -3.67 | -3.19 | 124.83 | 124.27 | 5.90 | 6.46 |
| C6 | 132.05 | 138.14 | 137.83 | -6.09 | -5.78 | 131.73 | 131.34 | 0.32 | 0.71 |
| C7 | 130.21 | 132.43 | 133.8 | -2.22 | -3.59 | 124.57 | 125.8 | 5.64 | 4.41 |
| C8 | 131.52 | 135.01 | 136.61 | -3.49 | -5.09 | 130.4 | 131.91 | 1.12 | -0.39 |
| C9 | 126.76 | 129.91 | 130.34 | -3.15 | -3.58 | 123.67 | 124.02 | 3.09 | 2.74 |
| C10 | 5.46 | 16.52 | 16.55 | -11.06 | -11.09 | 1.7 | 1.81 | 3.76 | 3.65 |
| C11 | 6.38 | 20.52 | 20.72 | -14.14 | -14.34 | 4.07 | 4.44 | 2.31 | 1.94 |
| C12 | 6.38 | 22.43 | 22.66 | -16.05 | -16.28 | 5.38 | 5.22 | 1.00 | 1.16 |
| C13 | 167.75 | 167.95 | 169.2 | -0.20 | -1.45 | 157.52 | 158.95 | 10.23 | 8.80 |
| H14 | 11.78 | 7.84 | 8.35 | 3.94 | 3.43 | 7.1 | 7.56 | 4.68 | 4.22 |
| H15 | 10.40 | 12.01 | 12 | -1.61 | -1.60 | 10.92 | 10.87 | -0.52 | -0.47 |
| H16 | 8.03 | 9.28 | 9.25 | -1.25 | -1.22 | 9.13 | 9.11 | -1.10 | -1.08 |
| H17 | 7.63 | 8.36 | 8.62 | -0.73 | -0.99 | 8.09 | 8.37 | -0.46 | -0.74 |
| H18 | 7.65 | 8.42 | 8.71 | -0.77 | -1.06 | 8.23 | 8.54 | -0.58 | -0.89 |
| H19 | 7.92 | 9.24 | 9.35 | -1.32 | -1.43 | 8.55 | 8.69 | -0.63 | -0.77 |
| H20 | 2.12 | 3.17 | 3.25 | -1.05 | -1.13 | 2.16 | 2.29 | -0.04 | -0.17 |
| H21 | 0.96 | 2.01 | 1.93 | -1.05 | -0.97 | 1.64 | 1.5 | -0.68 | -0.54 |
| H22 | 0.96 | 1.85 | 2.08 | -0.89 | -1.12 | 0.94 | 1.16 | 0.02 | -0.20 |
| H23 | 0.96 | 1.82 | 1.78 | -0.86 | -0.82 | 0.75 | 0.79 | 0.21 | 0.17 |
| H24 | 0.96 | 1.88 | 2.09 | -0.92 | -1.13 | 1.01 | 1.2 | -0.05 | -0.24 |
| H25 | 11.78 | 7.06 | 7.6 | 4.72 | 4.18 | 6.2 | 6.75 | 5.58 | 5.03 |
Table 4: The calculated 1H and 13C-NMR isotropic chemical shifts of title compound (with respect to TMS, all values in ppm) (6-31G(d,p))
| S. No. | Vibration Frequencies | HF | B3LYP |
|---|---|---|---|
| 1 | τ NCCC (18), τ CCCN(28) | 11 | 13 |
| 2 | τ NCCC(41), τ CCCN(25) | 21 | 29 |
| 3 | τ OCCC(45) | 39 | 39 |
| 4 | τ NNCN(13), τ NCNN(31), | 57 | 55 |
| 5 | δ NCC(15) | 67 | 71 |
| 6 | τ CNNC(39) | 100 | 94 |
| 7 | δ CCC(11), δ CCN(11), τ OCCC(11), τ CCCC(12) | 116 | 121 |
| 8 | τ CNNC(17) | 139 | 137 |
| 9 | δ CCN(21), τ CNNC(12), τ CCCC(12) | 152 | 166 |
| 10 | δ CNN(13), τ CCCC(17) | 202 | 202 |
| 11 | δ NCC(10), δ CCC(21), τ CCCN(11) | 211 | 217 |
| 12 | τ CNNC(32) | 227 | 236 |
| 13 | τ NCNN(21), τ CCCC(10) | 249 | 263 |
| 14 | δ CCC(17), δ OCC(29) | 279 | 284 |
| 15 | τ NCNN(11), τ NNCN(28), τ CCCC(18) | 323 | 334 |
| 16 | δ CCC(10) | 360 | 385 |
| 17 | ν CC(13), δ CCC(13), δ OCO(11) | 377 | 389 |
| 18 | τ CCCN(12), τ HNNC(10), τ NNCN(30) | 379 | 396 |
| 19 | δ NNC(10), τ CCCC(24) | 412 | 435 |
| 20 | δ CCC(11), τ CCCC(21) | 443 | 466 |
| 21 | τ HNNC(54) | 470 | 491 |
| 22 | δ OCC(17), τ CCCC(14) | 516 | 533 |
| 23 | δ OCC(10), τ CCCC(14), τ HCCC(10) | 543 | 570 |
| 24 | δ CCC(19) | 581 | 598 |
| 25 | τ HOCC(75) | 604 | 608 |
| 26 | δ OCN(17), δ CCC(16) | 620 | 644 |
| 27 | δ CCC(26), δ OCO(36) | 632 | 661 |
| 28 | ν CC(12), δ CCN(18) | 671 | 684 |
| 29 | τ CNNC(17), τ ONNC(15) | 694 | 748 |
| 30 | τ CCCC(29), τ ONNC(30) | 715 | 751 |
| 31 | τ NNCN(15), τ ONNC(39) | 722 | 774 |
| 32 | ν CC(12), δ OCO(11), δ CCC(16) | 745 | 801 |
| 33 | τ CCCC(12), τ HCCC(47) | 765 | 808 |
| 34 | ν NC(11), δ CNN(26) | 787 | 821 |
| 35 | δ HCC(46) | 797 | 823 |
| 36 | τ HCCC(28), τ ONNC(20) | 802 | 852 |
| 37 | δ CCC(17), τ HCCN(36) | 821 | 859 |
| 38 | ν NN(14), δ CCC(17), δ NCN(10) | 824 | 863 |
| 39 | ν CC(12), δ NNC(19), δ NCC(13), δ CCC(13) | 877 | 915 |
| 40 | ν CC(33), δ CCC(12), τ HCCC(11) | 897 | 931 |
| 41 | τ HCCC(69) | 903 | 965 |
| 42 | ν CC(28), δ CCC(15), τ HCCN(35) | 909 | 978 |
| 43 | τ HCCC(87) | 978 | 1054 |
| 44 | τ CCCC(13), τ HCNN(26), τ HCCC(44) | 996 | 1078 |
| 45 | τ HCNN(59), τ HCCC(20) | 1003 | 1081 |
| 46 | ν CC(10), δ NNC(22) | 1026 | 1087 |
| 47 | ν NN(10), τ HCCN(36) | 1054 | 1092 |
| 48 | ν CC(46), δ HCC(17) | 1060 | 1115 |
| 49 | τ HCCC(33), τ HCCN(10) | 1070 | 1133 |
| 50 | τ HCCC(28), τ HCCN(45) | 1083 | 1136 |
| 51 | ν CC(10), δ CCC(25), δ HCC(16) | 1095 | 1161 |
| 52 | τ HCCN(33) | 1125 | 1164 |
| 53 | ν OC(30), δ HCC(15), ν CC(11) | 1127 | 1181 |
| 54 | ν NN(24), τ HCCN(13) | 1136 | 1183 |
| 55 | ν CC(10), δ HCC(76) | 1180 | 1192 |
| 56 | ν CC(10), δ HOC(41) | 1198 | 1248 |
| 57 | δ HCC(72) | 1202 | 1251 |
| 58 | ν NC(18), δ HCC(13) | 1203 | 1255 |
| 59 | ν CC(53) | 1215 | 1264 |
| 60 | ν CC(12), ν NC(10) | 1235 | 1279 |
| 61 | ν NN(10), δ CNN(11) | 1271 | 1292 |
| 62 | δ HCC(40) | 1291 | 1343 |
| 63 | ν NC(18), ν NN(10), δ HCN(16) | 1326 | 1358 |
| 64 | ν CC(49) | 1339 | 1420 |
| 65 | ν OC(20), ν CC(11), δ HOC(26), δ OCO(14) | 1370 | 1440 |
| 66 | ν NC(12), δ HNN(65) | 1383 | 1471 |
| 67 | δ HCN(52) | 1421 | 1484 |
| 68 | δ HCH(97) | 1462 | 1521 |
| 69 | δ HCC(18), δ HCH(16) | 1472 | 1535 |
| 70 | ν CC(11), δ HCC(23), δ HCH(12) | 1475 | 1537 |
| 71 | ν CC(13), δ HCC(11), δ HCH(45) | 1506 | 1577 |
| 72 | δ HCC(25), δ HCH(13), δ CCC(13) | 1513 | 1587 |
| 73 | ν CC(29), δ CCC(10) | 1600 | 1681 |
| 74 | ν NC(45) | 1624 | 1716 |
| 75 | ν NC(31), ν CC(12) | 1633 | 1774 |
| 76 | ν NC(39), ν CC(16) | 1655 | 1810 |
| 77 | ν OC(44) | 1794 | 1878 |
| 78 | ν OC(39), ν NC(12) | 1808 | 1909 |
| 79 | ν CH(58) | 3117 | 3140 |
| 80 | ν CH(56) | 3120 | 3149 |
| 81 | ν CH(30) | 3159 | 3190 |
| 82 | ν CH(61) | 3175 | 3207 |
| 83 | ν CH(39) | 3188 | 3208 |
| 84 | ν CH(58) | 3194 | 3222 |
| 85 | ν CH(55) | 3197 | 3232 |
| 86 | ν CH(53) | 3203 | 3236 |
| 87 | ν CH(26) | 3212 | 3238 |
| 88 | ν CH(27) | 3217 | 3274 |
| 89 | ν NH(100) | 3664 | 3763 |
| 90 | ν OH(100) | 3720 | 3926 |
Table 5: The calculated IR frequencies of title compound (6-31G(d,p))
| Experimental Vibrations | IR (cm-1) |
|---|---|
| ν OC | 1263 |
| ν C=C | 1591 |
| ν C=N | 1607 |
| ν C=O | 1683 |
| ν C=O | 1698 |
| ν =CH | 3057 |
| ν NH | 3125 |
| ν OH | 3178 |
Table 6: The experimental IR frequencies of title compound
| Experimental (nm) | λ (nm) HF/B3LYP 6-31G(d,p) | Excitation Energy (eV) HF/B3LYP 6-31G(d,p) | f (osillatör strengths) HF/B3LYP 6-31G(d,p) |
|---|---|---|---|
| 308 | 235.61/335.03 | 5.2622/3.7007 | 0.5106/0.1245 |
| 262 | 230.87/316.19 | 5.3702/3.9211 | 0.1272/0.1771 |
| 232 | 216.64/303.51 | 5.7232/4.0850 | 0.0467/0.0010 |
Table 7: The experimental and calculated UV-vis values (HF/B3LYP 6-31G(d,p)) of title compound
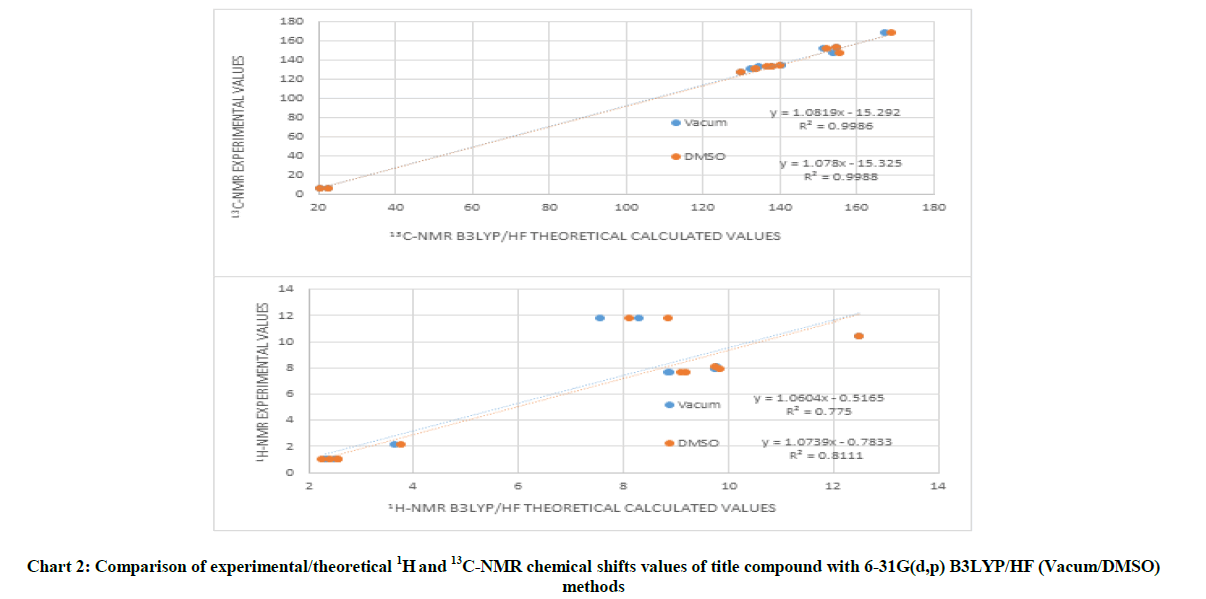
Chart 2: Comparison of experimental/theoretical 1H and 13C-NMR chemical shifts values of title compound with 6-31G(d,p) B3LYP/HF (Vacum/DMSO) methods
The GIAO 1H and 13C chemical shift values in gas phase/DMSO solvent (with respect to TMS) of title compound were calculated using the DFT (B3LYP) and Hartree Fock (HF) methods with 6-31G(d,p) basis set. The calculated and experimental data are shown in Table 4.
The calculated 1H chemical shift values are observed to be 0.94-10.92/1.82-12.01 ppm in gas phase and 0.79-10.87/1.78-12.00 in DMSO solvent at DFT (B3LYP)/HF methods, while the experimental parameters are calculated 0.96-11.78 ppm with 6-31G(d,p) basis set. Aromatic C-H signals were observed at 7.63-8.03 ppm. These signals were calculated 8.09-9.13/8.36-9.28 in gas phase and 8.37-9.11/8.62-9.25 ppm in DMSO solvent at B3LYP/HF levels. The cyclopropyl protons of this compound were observed at 0.96 ppm. These were calculated as 0.72-1.64/1.82-2.01 ppm in gas phase and 0.79-1.50/1.78-2.09 ppm in DMSO solvent at B3LYP/HF levels. The O-H signal was observed at 11.78 ppm. This signal was calculated as 6.20/7.06 ppm at gas phase and 6.75/7.60 ppm in DMSO solvent at B3LYP/HF levels. The N-H signal was observed at 11.78 ppm but these signals were observed at 7.1/7.84 ppm in gas phase and 7.56-7.84 ppm in DMSO solvent at B3LYP/HF levels. There is a slight difference between theoretical and experimental N-H signal because N-H proton of 4,5-dihydro-1H-1,2,4-triazol-5-one ring was displayed the acidic character.
The 13C-NMR chemical shift values of the title compounds are observed to be 1.70-157.72/20.52-167.95 ppm in gas phase and 1.81-158.95/20.72-169.20 in DMSO solvent at DFT (B3LYP)/HF methods, while the experimental parameters are calculated 5.46-167.75 ppm with 6-31G(d,p) basis set. As a result of, the R2 values of title compound were evaluated and 13C and 1H-NMR chemical shift values of title compound were plotted graphs (Chart 2). Theoretical and experimental between 13C and 1H-NMR chemical shifts ratios of title compound were observed a linear correlation. It is such a relationship between R-values of title compound; B3LYP(DFT)/HF 6-31G(d,p)Vacum:1H: 0.7750, 13C: 0.9986, B3LYP(DFT)/HF 6-31G(d,p)DMSO: 1H: 0.8111 13C: 0.9988.
The experimental O-H, N-H, C-H, C=O stretching vibrations were given in Table 6. There is a slight difference between theoretical vibrational and experimental frequencies. Theoretical vibrational frequencies for title compound are generally closer to the experimental frequencies.
The experimental absorption wavelenghts in ethanol solvent of title compound were recorded as 308, 262, 232 nm respectively. The excitation energies, oscillator strengths (ƒ) and absorption wavelengths (λ) of UV–vis electron absorption spectroscopy in ethanol solvent of title molecule have been calculated by using B3LYP/HF methods with 6-31G(d,p) basis set and were given in Figure 3 and Table 7. The calculated UV-vis values of title compound are in a very good agreement with the experimental values.
Electronic and thermodynamics properties, homo-lumo energies
The energy levels and distributions of HOMO and LUMO were calculated at DFT(B3LYP)/HF 6-31G(d,p) level for title compound. Energies of HOMO, LUMO were recorded as -7.649 -5.275 and 7.852 -5.201 eV B3LYP and HF/6-31G(d,p), respectively. Also, The molecular softness (S), hardness (η), electronegativity (χ), chemical potential (Pi), ionization potential (I), electron affinity (A), electrophilic (ω) and nucleophilic index (IP) were calculated by using HOMO and LUMO energies (Table 8). The ΔEH-L (energy gap) energies are 2.235/2.651 eV at B3LYP/HF level, respectively (Figure 4).
| B3LYP (6-31G(d,p)) | Hartree | Ev | Kcal/mol | Kj/mol | |
|---|---|---|---|---|---|
| LUMO | -0.194 | -5.275 | -121.635 | -508.927 | |
| HOMO | -0.281 | -7.649 | -176.404 | -738.083 | |
| A | Electron Affinity | 0.194 | 5.275 | 121.635 | 508.927 |
| I | Ionization Potential | 0.281 | 7.649 | 176.404 | 738.083 |
| ΔE | Energy Gap | 0.087 | 2.375 | 54.769 | 229.156 |
| χ | Electronegativity | 0.237 | 6.462 | 149.020 | 623.505 |
| Pi | Chemical Potential | -0.237 | -6.462 | -149.020 | -623.505 |
| ω | Electrophilic Index | 0.001 | 0.033 | 0.772 | 3.231 |
| IP | Nucleophilic Index | -0.010 | -0.282 | -6.503 | -27.210 |
| S | Molecular Softness | 22.914 | 623.519 | 14.378.914 | 60.162.005 |
| η | Molecular Hardness | 0.044 | 1.187 | 27.385 | 114.578 |
| HF (6-31G(d,p)) | Hartree | Ev | Kcal/mol | Kj/mol | |
| LUMO | -0.191 | -5.201 | -119.947 | -501.864 | |
| HOMO | -0.289 | -7.852 | -181.072 | -757.614 | |
| A | Electron Affinity | 0.191 | 5.201 | 119.947 | 501.864 |
| I | Ionization Potential | 0.289 | 7.852 | 181.072 | 757.614 |
| ΔE | Energy Gap | 0.097 | 2.651 | 61.125 | 255.750 |
| χ | Electronegativity | 0.240 | 6.527 | 150.510 | 629.739 |
| Pi | Chemical Potential | -0.240 | -6.527 | -150.510 | -629.739 |
| ω | Electrophilic Index | 0.001 | 0.038 | 0.879 | 3.678 |
| IP | Nucleophilic Index | -0.012 | -0.318 | -7.331 | -30.671 |
| S | Molecular Softness | 20.532 | 558.684 | 12.883.749 | 53.906.170 |
| η | Molecular Hardness | 0.049 | 1.325 | 30.563 | 127.875 |
Table 8: The calculated electronic properties of title compound (6-31G(d,p) B3LYP/HF)
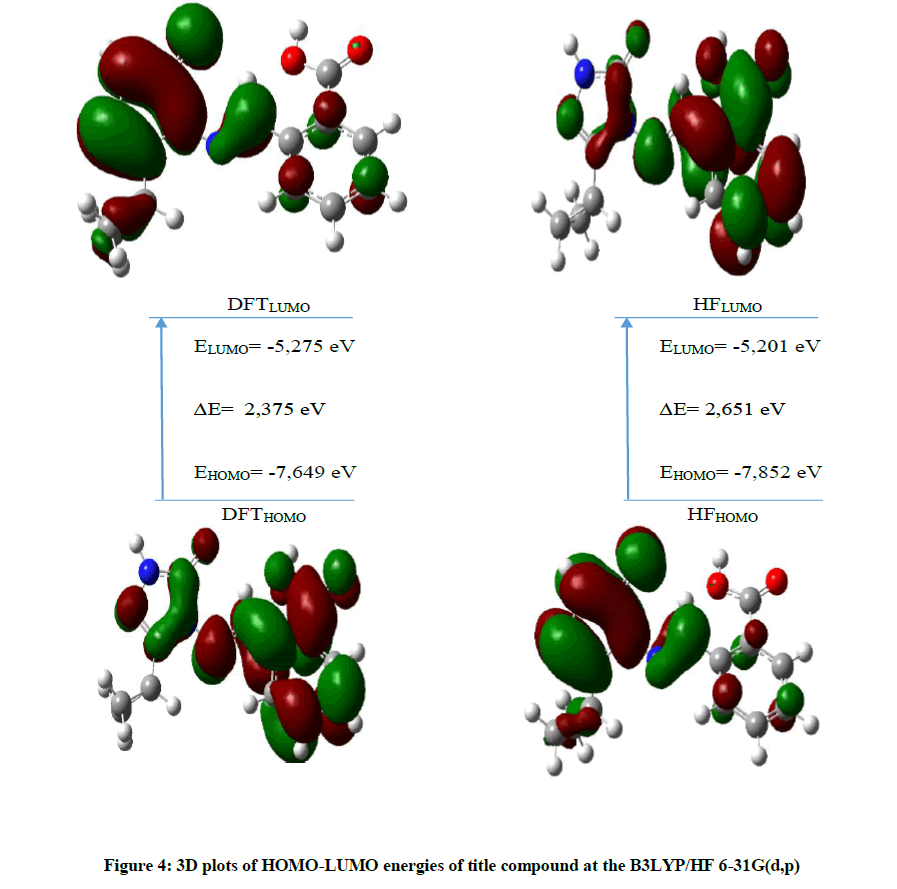
Figure 4: 3D plots of HOMO-LUMO energies of title compound at the B3LYP/HF 6-31G(d,p)
The dipole moment values of title compound were computed as 0.581/0.606 D for DFT(B3LYP)/HF methods with the 6-31G(d,p) basis set. The results were given in Table 9. The highest dipole moment is recorded as μz component. The mean polizability (<α>) and the mean first-order hyperpolarizability (<β>) and are calculated -47.160 × 10-24 esu/23,088 × 10-24 esu and 8.107 × 10-30 esu/3.169 × 10-30 esu for B3LYP/HF, respectively. It was found that the β values at B3LYP/HF methods of title compound is higher than that of urea.
| B3LYP (6-31G(d,p)) | HF(6-31G(d,p)) | |
|---|---|---|
| µx | -1.558 Debye | 1.141 Debye |
| µy | 2.599 Debye | 3.240 Debye |
| µz | 0.581 Debye | 0.606 Debye |
| µToplam | 3.085 Debye | 3.488 Debye |
| αxx | -116.52 a,u, | 31.522 a.u. |
| αyy | -26.19 a.u. | 24.379 a.u. |
| αzz | 1.24 a.u. | 13.362 a.u. |
| Α | -47.160 × 10-24 esu | 23.088 × 10-24 esu |
| ∆α | 106.71810-24 esu | 15.846 × 10-24 esu |
| Βx | -7850.88 a.u. | 3071.51 a.u. |
| βy | 1966.29 a.u. | 409.35 a.u. |
| βz | -463.44 a.u. | -663.47 a.u. |
| βxxx | -2361.58 a.u. | 189.72 a.u. |
| βxxy | -125.85 a.u. | 89.16 a.u. |
| βxyy | -9.83 a.u. | 189.72 a.u. |
| βyyy | -137.92 a.u. | -20.12 a.u. |
| βxxz | 76.32 a.u. | 6.64 a.u. |
| βxyz | -13.01 a.u. | 9.71 a.u. |
| βyyz | 17.39 a.u. | -40.84 a.u. |
| βxzz | -90.96 a.u. | -12.44 a.u. |
| βyzz | -40.22 a.u. | -20.12 a.u. |
| βzzz | 62.80 a.u. | -45.08 a.u. |
| β | 8.107 × 10-30 esu | 3.169 × 10-30 esu |
Table 9: The mean polizability (<α>), the anisotropy of the polarizability (Δα), the mean first-order hyperpolarizability (<β>), dipole moment values of title compound
The MEP map of title compound is shown in Figure 5. The MEP map shows that the positive potential sites are around the hydrogen as well as the negative potential sites on electronegative oxygen, nitrogen atoms. Also, thermodynamic properties of title compound are given in Table 10.
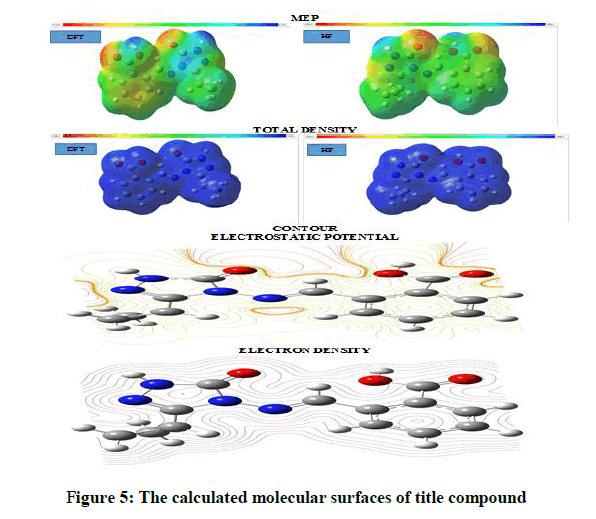
Figure 5: The calculated molecular surfaces of title compound
| Rotational temperatures (Kelvin) | DFT | HF |
|---|---|---|
| A | 0.02847 | 0.02796 |
| B | 0.00917 | 0.00936 |
| C | 0.00721 | 0.00773 |
| Rotational constants (GHZ) | ||
| A | 0.59328 | 0.5826 |
| B | 0.19116 | 0.19497 |
| C | 0.1503 | 0.16112 |
| Zero-point vibrational energy (Kcal/Mol) | 153.21673 | 165.36306 |
| Thermal correction to Energy | 0.261506 | 0.279787 |
| Thermal correction to Enthalpy | 0.26245 | 0.280731 |
| Thermal correction to Gibbs Free Energy | 0.195555 | 0.216273 |
| Sum of electronic and zero-point Energies | -947.016165 | -941.378701 |
| Sum of electronic and thermal Energies | -946.998826 | -941.362437 |
| Sum of electronic and thermal Enthalpies | 946.997882 | -941.361493 |
| Sum of electronic and thermal Free Energies | -947.064777 | 941.42595 |
| Thermal Energies E (Kcal/mol) | ||
| Translational | 0.889 | 0.889 |
| Rotational | 0.889 | 0.889 |
| Vibrational | 162.320 | 173.791 |
| Total | 164.097 | 175.569 |
| Thermal Capacity CV (Cal/Mol-Kelvin) | ||
| Translational | 2.981 | 2.981 |
| Rotational | 2.981 | 2.981 |
| Vibrational | 59.289 | 54.379 |
| Total | 65.251 | 60.34 |
| Entropy S (Cal/Mol-Kelvin) | ||
| Translational | 42.702 | 42.702 |
| Rotational | 34.199 | 34.129 |
| Vibrational | 63.891 | 58.833 |
| Total | 140.792 | 135.663 |
Table 10: The calculated thermodynamics parameters of title compound (6-31G(d,p) B3LYP/HF)
Conclusion
In this study, the geometric, thermodynamics, spectroscopic and electronic values of title compound have been calculated by using DFT (B3LYP) and HF methods with the 6-31G(d,p) basis sets. The UV-vis, IR, 1H- and 13C- NMR spectra have been obtained and compared with the experimental spectroscopic values. The theoretical calculated spectra values were found good agreement with experimental UV-vis, IR, 1H- and 13C- NMR spectra values. The value of the energy gap between the HOMO-LUMO energies was determined. Geometric parameters; bond angels and bond lengths were compared with experimental values obtained from literature [33,34]. All results showed that the calculated spectroscopic, geometric, thermodynamics and electronic parameters obtained by B3LYP/6-31G(d,p) method had a better agreement with the experimental values than HF/6-31G(d,p) method.
References
- B. Tozkoparan, E. Küpeli, E. Yeşilada, M. Ertan, Bioorg. Med. Chem., 2007, 15, 1808-1814.
- K. Sancak, Y. Ünver, D. Ünlüer, E. Düğdü, G. Kör, F. Çelik, E. Birinci, Turk. J. Chem., 2012, 36, 457-466.
- Y. Ünver, K. Sancak, F. Çelik, E. Birinci, M. Küçük, S. Soylu, N.A. Burnaz, Eur. J. Med. Chem., 2014, 84, 639-650.
- D.J.V.C. Steenis, O.R.P. David, G.P.F. Strijdonck, J.H. Maarseveen, J.N.H. Reek, Chem. Commun., 2005, 34, 4333-4335.
- V.V. Rostovtsev, L.G. Green, V.V. Fokin, K.B. Sharpless, Angew. Chem. Int. Ed., 2002, 41, 2596-2599.
- F. Himo, T. Lovell, R. Hilgraf, V.V. Rostovtsev, L. Noodleman, K.B. Sharpless, V.V. Fokin, J. Am. Chem. Soc., 2005, 127, 210-216.
- A.J. Dirks, S.S. Berkel, N.S. Hatzakis, J.A. Opsteen, F.L. Delft, J.J.L.M. Cornelissen, A.E. Rowan, J.C.M. Hest, F.P.J.T. Rutjes, R.J.M. Nolte, Chem. Commun., 2005, 33, 4172-4174.
- M. Meldal, C.W. Tornoe, Chem. Rev., 2008, 108, 2952-3015.
- Z. Ya-Bin, Y. Ze-Yi, L. Yong-Min, Tetrahedron Lett., 2006, 47, 1545-1549.
- S. Saqlain Haider, M.S. Alam, H. Hamid, S. Shafi, A. Nargotra, P. Mahajan, S. Nazreen, M.K. Arunasree, C. Kharbanda, Y. Ali, A. Alam, K.P. Amulya, Eur. J. Med. Chem., 2013, 7, 579-588.
- B. Garudachari, A.M. Isloor, M.N. Satyanarayana, F. Hoong-Kun, G. Hegde, Eur. J. Med. Chem., 2014, 74, 324-332.
- A. Lauria, C. Patella, P. Diana, P. Barraja, A. Montalbano, G. Cirrincione, G. Dattolo, A.M. Almerico, Tetrahedron Lett., 2006, 47, 2187-2190.
- M. Pagacz-Kostrzewa, R. Bronisz, M. Wierzejewska, Chem. Phy. Lett., 2009, 473, 238–246.
- M. ElBelghiti, Y. Karzazi, A. Dafali, B. Hammouti, F. Bentiss, I.B. Obot, I. Bahadur, E.E. Ebenso, J. Mol. Liq., 2016, 218, 281–293.
- J.W. Wang, L.Y. Ding, B.Q. Wang, Y.Y. He, Y. Guo, X.F. Jia, W. Zheng, J. Mol. Struct., 2014, 1058, 62-70.
- R.Y. Jin, X.H. Sun, Y.F. Liu, W. Long, W.T. Lu, H.X. Ma, J. Mol.Struct., 2014, 1062, 13-20.
- A.S. Al-Tamimi, J. Mol. Struct., 2016, 1120, 215-227.
- N. Süleymanoğlu, Y. Ünver, R. Ustabaş¸ S. Direkel, G. Alpaslan, J. Mol. Struct., 2017, 1144, 80-86.
- S. Çelik, S. Badoğlu, Ş. Yurdakul, Vib. Spectrosc., 2017, 92, 280-286.
- R.Y. Jin, C.Y. Zeng, X.H. Liang, X.H. Sun, Y.F. Liu, Y.Y. Wang, S. Zhou, Bioorg. Chem., 2018, 80, 253-260.
- T. Karakurt, M. Dinçer, A. Çetin, M. Şekerci, Spectra. Chim. Acta A., 2010, 77, 189-198.
- T. Tian, X. Hu, P. Guan, S. Wang, X. Ding, J. Mol. Liq., 2017, 248, 70-80.
- W. Kowhakul, D. Inoue, Y. Nakagawa, H. Masamoto, M. Shigematsu, Journal of Loss Prevention in the Process Industries, 2017, 50, 37-54.
- L. Guennouna, J. El jastimi, F. Guedira, K. Marakchi, O.K. Kabbaj, A. El Hajji, S. Zaydoun, Spectra. Chim. Acta Part A., 2011, 78, 347-353.
- R. John Xavier, E. Gobinath, Spectra. Chim. Acta A., 2012, 86, 242-251.
- V. Karunakaran, V. Balachandran, Spectrochim. Acta A., 2012, 98, 229-239.
- A. Suvitha, S. Periandy, S. Boomadevi, M. Govindarajan, Spectrochim. Acta A, 2014, 117, 216-224.
- M.H. Jamróz, Vibrational energy distribution analysis: VEDA 4 program, 2004, Warsaw.
- M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G.A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H.P. Hratchian, A.F. Izmaylov, J. Bloino, G. Zheng, J.L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J.A. Montgomery Jr., J.E. Peralta, F. Ogliaro, M. Bearpark, J.J. Heyd, E. Brothers, K.N. Kudin, V.N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J.C. Burant, S.S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N.J. Millam, M. Klene, J.E. Knox, J.B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R.E. Stratmann, O. Yazyev, A.J. Austin, R. Cammi, C. Pomelli, J.W. Ochterski, R.L. Martin, K. Morokuma, V.G. Zakrzewski, G.A. Voth, P. Salvador, J.J. Dannenberg, S. Dapprich, A.D. Daniels, O. Farkas, J.B. Foresman, J.V. Ortiz, J. Cioslowski, D.J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2009.
- R. Dennington, T. Keith, J. Millam, GAUSSVIEW, Version 5, Semichem Inc., Shawnee Mission, KS, 2009.
- R. Ditchfield, J. Chem. Phys., 1972, 56, 5688-5691.
- K. Wolinski, J.F. Hinton, P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251-8260.
- R.J. Fessenden, J.S. Fessenden, Organic Chemistry, Third Edition, Brooks, Cole Publishing Company, California, 1986.
- A.A. İkizler, Organik Kimyaya Giriş, Dördüncü Baskı, KTÜ Basımevi, Trabzon, Türkiye, 1996, 398s.



