Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 5
Synthesis and Biological Evaluation of Certain New Benzimidazole Derivatives as Cytotoxic Agents New Cytotoxic Benzimidazoles
Lamia W Mohamed1*, Azza T. Taher1,2, Ghada S Rady3, Mamdouh M Ali4 and Abeer E Mahmoud4
1Pharmaceutical Organic Chemistry Department, Faculty of Pharmacy, Cairo University, Cairo, 11562, Egypt
2Pharmaceutical Organic Chemistry Department, Faculty of Pharmacy, October 6 University, 6th October City, Giza, Egypt
3Directorate of Health Affairs, Ministry of health, Giza, 11562, Egypt
4Biochemistry Department, Division of Genetic Engineering and Biotechnology, National Research Centre, Giza, Egypt
- *Corresponding Author:
- Lamia W Mohamed
Pharmaceutical Organic Chemistry Department
Faculty of Pharmacy
Cairo University
Cairo, 11562, Egypt
Abstract
Novel series of benzimidazole has been synthesizedand evaluated for their anti-proliferative activity against human breast adenocarcinoma MCF-7, human lung carcinoma A549 and human epitheloid cervix carcinoma HELA. Some derivatives showed potent growth inhibitory activity, in particular compounds 4a, 4b, 5a and 7b. N-(1H-benzimidazo-2-yl)-2-(1H-benzimidazo-2-ylamino)acetamide 5a was found to be the most potent with percentage inhibition 95% on MCF-7 cell line and 77% on A549 cell line compared to 60% inhibition of cisplatin. The antitumor activity of 5a was accompanied by significant increase in the activity of superoxide dismutase with concomitant decrease in the activities of catalase, glutathione peroxidase and reduced glutathione level. Accordingly, the overproduction of hydrogen peroxide and nitric oxide allowed Reactive Oxygen Species (ROS)-mediated tumour cells death, as monitored by reduction in the synthesis of protein and nucleic acids.
Keywords
Benzimidazole, MCF-7, Anticancer, Antiproliferative, Cytotoxic, Antioxidant, ROS
Introduction
Cancer is a serious worldwide health threat, killing almost seven million people a year. They account for about one-fifth of all deaths worldwide [1]. Among all types of cancer, lung, breast, colorectal, stomach and prostate cancers are the underlying causes for the majority of cancer deaths [2]. Furthermore, breast carcinoma is the most common cancer in women worldwide and remains the most frequent cause of malignancy associated deaths among women [3]. Excessive unbalanced production of Reactive Oxygen Species (ROS) has been well established to be associated with many pathological conditions including cancer and inflammation [4].
Benzimidazole is an important pharmacophore building block providing diversity of applications. It acts as proton pump inhibitor [5], antihypertensive [6], anti-inflammatory [7] and anti-psychotic [8]. We were concerned in its anticancer effect, which was revealed by pyrimidobenzimidazole derivatives i, iia, b [9,10] and pyrazolobenzimidazoles iii, iva, b, v [10-12]. The benzimidazole dimers possessed promising anticancer effect as compound vi which showed DNA intercalation, while Hoechst 33258 and Hoechst 33342 viia, b exhibited topoisomerase inhibiting effect [13,14]. The anthraquinone moiety was contained in anticancer drugs as doxorubicin and daunorubicin viii a-b. Similarly, compound ix was active anticancer derivative against MCF-7 cell line [15] (Figure 1).
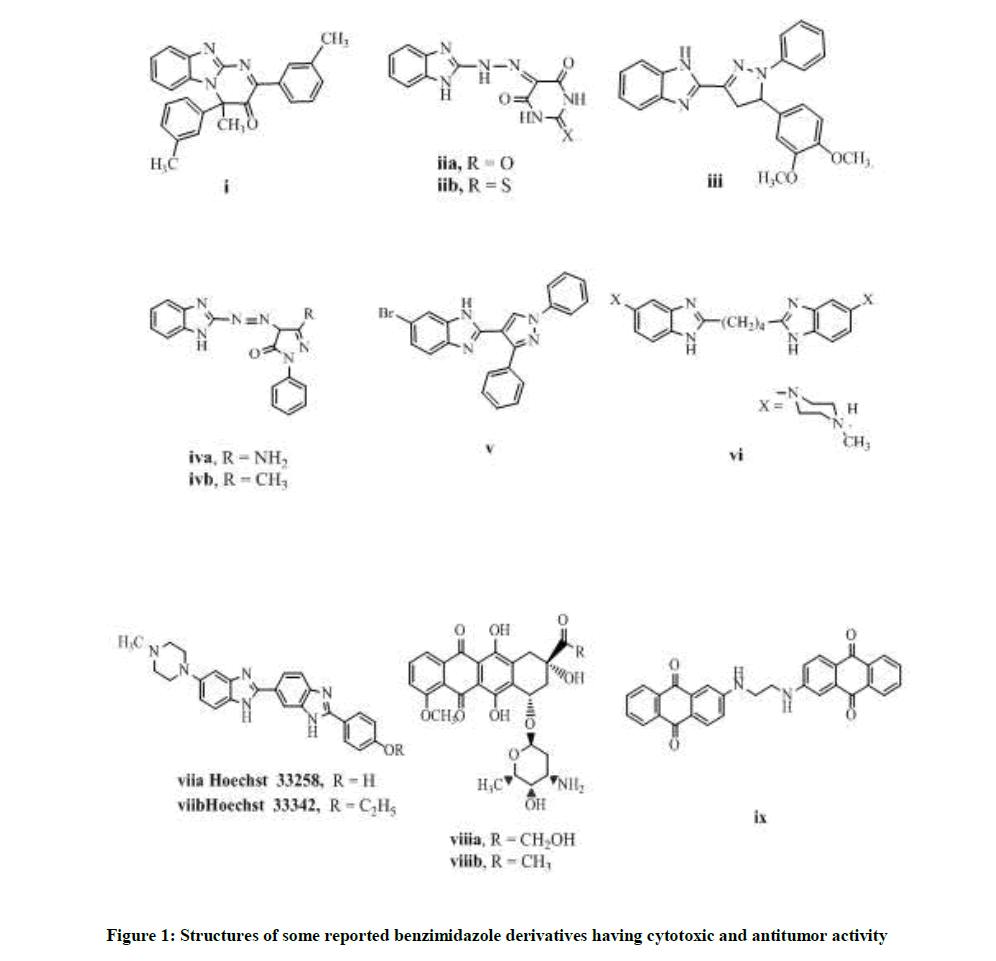
Figure 1: Structures of some reported benzimidazole derivatives having cytotoxic and antitumor activity
Materials and Methods
Chemical Synthesis
All reagents were commercially available and were used without further purification. Melting points were recorded on a Griffin apparatus and are uncorrected. Microanalyses for C, H and N were carried out at the micro-analytical center, Al-Azhar University, Egypt. Infrared Radiation (IR) spectra were recorded on Shimadzu IR 435 spectrophotometer in KBr disc, Faculty of pharmacy, Cairo University, Egypt and values were represented in ʋ cm-1. Proton Nuclear Magnetic Resonance (1H-NMR) spectra were obtained on a Mercury-300 BB MHz spectrophotometers using TMS as an internal reference, shift values were recorded in ppm on δ scale, at Micro-analytical center, Cairo University, and Bruker Ascend 400 MHz, Faculty of pharmacy, Cairo University, Egypt. Carbon-13 Nuclear Magnetic Resonance (13C-NMR) spectra were recorded on Varian Gemini 100 MHZ spectrophotometer using TMS as internal standard and chemical shift values were recorded in ppm on δ scale, Main Defence Chemical Laboratory, El Khanka, Al-Qalyubiyah, Egypt. Mass spectra were run on GCMS-QP2010 Plus spectrometer, at Micro-analytical center, Cairo University, Egypt. Progress of the reaction and the purity of products were monitored by TLC pre-coated aluminum sheet silica gel (Merck 60F 254) and was visualized by UV lamp (C6H6/(CH3)2O 9:1) and (CHCl3/CH3OH 9:1).
2-Aminobenzimidazole (1) was purchased from Aldrich chemicals, 4-amino-1H-benzimidazo[1,2-a]pyrimidin-2-one (2) was synthesized according to reported procedure [16]. N-(1H-benzimidazol-2-yl)-2-chloroacetamide (8) was synthesized according to reported procedure [17- 21].
3-[2-(5-oxo-1-phenyl-4,5-Dihyodr-1H-pyrazol-3-yl)diazen-1-yl]-4-aminobenz-imidazo[1,2-a] pyrimidin-2(1H)-one (3)
This compound was synthesized when an ice-cold solution of 3-amino-1-phenyl-1H-pyrazol-5(4H)-one diazonium salt [prepared from 3-amino- 1-phenyl-1H-pyrazol-5(4H)-one (0.17 g, 0.10 mmol), concentrated hydrochloric acid (0.03 g, 0.10 mmol) and sodium nitrite (0.06 g, 0.10 mmol) in water (3 ml)] was added to a chilled solution of 2 (0.2 g, 0.10 mmol) and sodium hydroxide (0.16 g, 0.10 mmol) in water (3 ml). The reaction mixture was maintained at -5°C with continuous stirring for 30 min, then acidified with glacial acetic acid till pH 5-5.5. The resulting solid was filtered, washed with water, dried and crystallized from ethanol /acetonitrile (1:1).
Yield: 73.6%; M. P. > 300°C ; IR: (KBr) ν max: 3425, 3344, 3082, 2889, 1675-1670, 1639, 1612, 1546 cm-1; 1H-NMR (DMSO-d6, 300 MHz) δ (ppm)=4.12 (s, 2H, NH2, D2O exchangeable), 4.99 (s, 2H, pyrazole-C4-H), 7.06 (s, 1H, NH, D2O exchangeable), 7.1 (t, 1H, phenyl-C4-H), 7.19 (t, 2H, phenyl-C3,5-H), 7.43 (t, 2H, benzimidazole-C5,6-H), 7.48-7.51 (d, 2H, J=8.10 Hz, phenyl-C2,6-H), 8.03 (d, 2H, J=8.40 Hz, benzimidazole-C4,7-H); MS m/z (% rel. Int.): 388.35 (2.43%, M+2), 387.30 (11.13%, M+1), 386.30 (49.04%, M+.), 73 (100%, C3H7NO). Anal. Calcd. for C19H14N8O2 (386.37): C, 59.06, H, 3.65, N, 29.00. Found: C, 59.14, H, 3.64, N, 29.09.
General procedure for the synthesis of 4-[2-(substitued)diazen-1-yl]benzimidazo[1,2-a]pyrimidin-2(1H)-ones 4(a-d)
An ice-cold solution of 2 diazonium salt [prepared from 2 (0.20 g, 0.10 mmol), concentrated hydrochloric acid (0.03 g, 0.10 mmol) and sodium nitrite (0.06 g, 0.10 mmol) in water (3 ml)] was added to (0.10 mmol) chilled solution of either 3-amino-1-phenyl-1H-pyrazol-5(4H)-one (0.175 g), 3-methyl-1-phenyl-1H-pyrazol-5(4H)-one (0.17 g), pyrimidine-2,4,6 (1H, 3H, 5H)-trione (0.12 g) or dihyodr-2-thioxopyrimidine-4,6 (1H, 5H)-dione (0.14 g) and sodium hydroxide (0.16 g, 0.1 mmol) in water (3 ml). The reaction mixture was maintained at -5°C with continuous stirring for 30 min, and then acidified with glacial acetic acid till pH 5-5.5. The resulting solid was filtered, washed with water, dried and crystallized from ethanol/acetonitrile (1:1).
4-[2-(3-amino-1H-1-phenyl-5(4H)oxo-pyrazol-4-yl)diazen-1-yl]benzimidazo[1,2-a]pyrimidin-2(1H)-one (4a)
Yield: 90.60%; M. P. >300°C; IR (KBr) ν max: 3540-3300, 3040, 2990, 1697, 1685, 1635, 1573 cm-1; 1H-NMR (DMSO-d6, 300MHz) δ (ppm)= 4.06 (s, 1H, pyrazole-C4-H), 6.18 (s, 2H, NH2, D2O exchangeable), 6.51 (s, 1H, NH, D2O exchangeable), 6.78 (t, 1H, phenyl-C4-H), 6.94 (t, 2H, phenyl-C3,5-H), 7.03 (d, 2H, J=3.00 Hz, phenyl–C2,6-H), 7.27 (t, 2H, benzimidazole-C5,6-H), 7.99 (d, 2H, J=8.10 Hz, benzimidazole-C4,7-H), 8.49 (s, 1H, pyrimidinone-C5-H); MS m/z (% rel. Int.): 388.30 (3.17 %, M+ +2), 387.30 (6.61%, M+ +1), 386.30 (3.54 %, M+.), 71 (100%, C3H5NO). Anal. Calcd. for C19H14N8O2 (386.37): C, 59.06, H, 3.65, N, 29.00. Found: C, 59.13, H, 3.62, N, 29.13.
4-[2-(3-methyl-1H-1-phenyl-5(4H)oxo-pyrazol-4-yl)diazen-1-yl]benzimidazo[1,2-a]pyrimidin-2(1H)-one (4b)
Yield: 57.10%; M. P. 244-246°C; IR (KBr) ν max: 3414, 3078, 2989, 1690, 1681, 1647, 1612, 1570 cm-1; 1H-NMR (DMSO-d6, 300 MHz) δ (pppm)=2.07 (s, 3H, CH3), 5.00 (s, 1H, pyrazole-C4-H), 7.00 (s, 1H, NH, D2O exchangeable), 7.19 (t, 3H, phenyl-C3,4,5-H), 7.37 (t, 2H, benzimidazole-C5,6-H), 7.48 (d, 2H, J=7.80 Hz, phenyl-C2,6-H), 8.04 (d, 2H, J=8.10 Hz, benzimidazole-C4,7-H), 8.33 (s, 1H, pyrimidinone-C5- H); 13C NMR(DMSO-d6, 100 MHz) δ (ppm)=40.27 (CH3), 79.42 (pyrazole-C4), 112.58 (pyrimidinone-C5), 117.28 (benzimidazole-C4,7), 120.16 (phenyl-C2,6), 124.14 (phenyl-C4, benzimidazole-C5,6), 128.40 (phenyl-C3,5), 142.66 (benzimidazole-C1a), 147.70 (pyrimidinone-C4, benzimidazole-C3a, phenyl-C1), 151.48 (pyrimidinone-C2, pyrazole-C3), 161.99 (2C=O); MS m/z (% rel. Int.): 387.25 (7.71%, M++2), 386.25 (26.87%, M+ +1), 385.25 (1.21 %, M+.), 236.15 (100%, C12H20N4O). Anal. Calcd. for C20H15N7O2 (385.38): C, 62.33, H, 3.92, N, 25.44. Found: C, 62.41, H, 3.95, N, 25.52.
4-[2-(2,4,6(1H,3H,5H)-trioxo-pyrimidine-5-yl)diazen-1-yl]benzimidazo[1,2-a] pyrimidin-2(1H)-one (4c)
Yield: 73.70%; M. P: 272-274°C; IR (KBr) ν max: 3450-3360, 3078, 2985, 1720, 1700, 1680, 1674, 1612, 1550 cm-1; 1H-NMR (DMSO-d6, 300 MHz) δ (ppm)=4.99 (s, 1H, barbituric-C5-H), 7.07 (s, 1H, pyrimidinone-NH, D2O exchangeable), 7.14 (t, 1H, benzimidazole-C5-H), 7.27 (t, 1H, benzimidazole-C6-H), 7.49 (d, 1H, J=8.10 Hz, benzimidazole-C4-H), 8.04 (d, 1H, J=8.10 Hz, benzimidazole-C7-H), 8.48 (s, 1H, pyrimidinone- C5-H), 9.72 (brs, 1H, NH, D2O exchangeable), 10.24 (brs, 1H, NH, D2O exchangeable); MS m/z (% rel. Int.): 340.95 (1.65%, M+ +2), 340.00 (1.73%, M++1), 339.00 (3.77%, M+), 77 (100%, C2H11N3). Anal. Calcd. for C14H9N7O4 (339.27): C, 49.56, H, 2.67, N, 28.90. Found: C, 49. 62, H, 2.69, N, 28.99.
4-[2-(2(3H)thioxo-4,6(1H,5H)dioxo-pyrimidine-5-yl)diazen-1-yl]benzimidazo[1,2-a] pyrimidin-2(1H)-one (4d)
Yield: 61.90%; M. P. > 300°C; IR. (KBr) ν max: 3460-3400, 3174, 2889, 1705, 1674, 1639, 1608, 1562 cm-1; 1H-NMR (DMSO-d6, 300 MHz) δ (ppm)=5.03 (s, 1H, thiobarbituric-C5-H), 7.12 (t, 1H, benzimidazole-C5-H), 7.26 (t, 1H, benzimidazole-C6-H), 7.48 (d, 1H, benzimidazole-C4- H), 7.59 (1H, pyrimidinone-NH, D2O exchangeable), 8.06 (d, 1H, benzimidazole-C7-H), 8.31 (s, 1H, pyrimidinone-C5-H), 9.40 (brs, 1H, NH, D2O exchangeable), 10.32 (brs, 1H, NH, D2O exchangeable); MS m/z (% rel. Int.): 356.20 (3.84%, M++1), 355.20 (2.42%, M+.), 119.05 (100%, C4H11N2O2). Anal. Calcd. for C14H9N7O3S (355.33) C, 47.32, H, 2.55, N, 27.59. Found: C, 47.39, H, 2.58, N, 27.71.
General procedure for the synthesis of N-(benzi-midzol-2-yl)-2or3-(benzi-midzol-2ylamino)alkan-amides 5(a-c)
A mixture of 2-aminobenzimidazole (0.50 g, 0.36 mmol), appropriate acid chloride (0.18 mmol) [chloroacetyl chloride (0.21 g), 2- chloropropanoyl chloride (0.24 g), 3-chloropropanoyl chloride (0.23 g)], and anhydrous potassium carbonate (0.51 g, 0.37 mmol) in dry benzene (20 ml) was heated under reflux for (13-20) h. After cooling the solvent was evaporated under reduced pressure, ethanol (0.5 ml) in ice was added and stirred for half an hour. The separated solid was filtered, washed with water (2 x 20 ml), dried and crystallized from ethanol.
N-(1H-benzimidazo-2-yl)-2-(1H-benzimidazo-2-ylamino)acetamide (5a)
Yield: 49%; M. P. 102-104°C; IR (KBr) ν max: 3440-3317, 3062, 2960, 1685, 1631, 1558 cm-1; 1H-NMR (DMSO-d6, 300 MHz) δ (ppm)=4.37 (s, 2H, CH2-H), 7.05-7.18 (m, 4H, benzimidazole-C5,6-H), 7.08 (s, 1H, NH, D2O exchangeable), 7.14 (s, 1H, NH, D2O exchangeable), 7.39-7.42 (m, 4H, benzimidazole-C4,7-H), 7.45 (s, 1H, NH, D2O exchange- able), 7.6 (s, 1H, NH, D2O exchangeable); 13C-NMR(DMSO-d6, 100 MHz) δ (ppm)=46.60(CH2), 110.38 (benzimidazole -C4,7), 113.73 (benzimidazole-C5,6), 120.61 (benzimidazole-C3a,7a), 121.71 (benzimidazole-C2), 167.25 (C=O); MS (EI) m/z (% rel. Int.): 307.90 (0.22%, M++1), 306.90 (0.68%, M+.), 133.05 (100%, C7H7N3). Anal. Calcd. for C16H14N6O (306.32) C, 62.74, H, 4.61, N, 27.44. Found: C, 62.88, H, 4.58, N, 27.61.
N-(1H-benzimidazo-2-yl)-2-(1H-benzimidazo-2-ylamino)propanamide (5b)
Yield: 59%; M. P. 208-210°C; IR (KBr) ν max: 3400-3320, 3095, 2920, 1693, 1630, 1597 cm-1; 1H-NMR (DMSO-d6, 300 MHz) δ (ppm)= 1.57 (d, 3H, J=6.90 Hz, CH3), 3.34 (q, 1H, J=6.00 Hz, CH-H), 5.36-5.75 (br s, 2H, NH, D2O exchangeable), 6.95-6.99 (m, 4H, benzimidazole-C5.6H), 6.98 (s, 1H, NH, D2O exchangeable), 7.16 (s, 1H, NH, D2O exchangeable), 7.16-7.18 (m, 4H, benzimidazole-C4,7-H); 13C-NMR (DMSO-d6, 100 MHz) δ (ppm)=20.83(CH3), 54.51(CH), 113.75 (benzimidazole -C4,7), 119.81 (benzimidazole-C5,6), 120.53 (benzimidazole-C3a,7a), 121.40(benzimidazole-C2), 170.01 (C=O); MS m/z (% rel. Int.): 321.20 (1.36%, M++1), 320.20 (15.76%, M+.), 132.15 (36.72%), 51 (100%). Anal. Calcd. for C17H16N6O (320.35) C, 63.74, H, 5.03, N, 26.23. Found: C, 63.85, H, 5.07, N, 26.37.
N-(1H-benzimidazo-2-yl)-3-(1H-benzimidazo-2-ylamino)propanamide (5c)
Yield: 65.90%; M. P: 198-200°C; IR (KBr) ν max: 3400-3315 (4 NH), 3055, 2920, 1685, 1636, 1577 cm-1; 1H-NMR (DMSO-d6, 300 MHz): 2.88 (t, 2H, CH2-H, J=6.90, 7.2 Hz), 4.24 (t, 2H, CH2- H, J=6.90, 7.2 Hz), 7.07 (s, 1H, NH, D2O exchangeable), 7.11 (m, 4H,benzimidazole–C5,6 -H), 7.12 (s, 1H, NH, D2O exchangeable), 7.14 (s, 1H, NH, D2O exchangeable), 7.36 (s, H, NH, D2O exchangeable), 7.38 (m, 4H, benzimidazole-C4,7-H); 13C-NMR (DMSO-d6, 100 MHz) δ (ppm)= 29.91 (CH2), 36.56 (CH2), 111.42 (benzimidazole -C4,7), 117.10 (benzimidazole-C5,6), 120.57 (benzimidazole-C3a,7a), 121.23 (benzimidazole-C2), 168.17 (C=O); MS (EI) m/z (% rel. Int.): 321 (1.06%, M++1), 320 (0.89%, M+.), 133.05 (100%, C7H7N3). Anal. Calcd. for C17H16N6O (320.35) C, 63.74, H, 5.03, N, 26.23. Found: C, 63.60, H, 5.09, N, 26.37.
(1H-benzimidazol-2-ylcarbamoyl)formyl chloride (6)
A mixture of 2-aminobenzimidazole (2.00 g, 1.50 mmol), oxalyl dichloride (1.90 g, 1.50 mmol) and anhydrous potassium carbonate (2.07 g, 1.50 mmol) was heated under reflux for 4 h in dry benzene (20 ml). The separated solid was filtered, washed with water (2 x 20 ml), dried and crystallized from ethanol.
Yield: 76.64%; M. P. 250-252°C; IR (KBr) ν max: 3442, 3250, 3115, 1724, 1681, 1624, 1591 cm-1; 1H-NMR (DMSO-d6, 400 MHz) δ (ppm)= 7.43 (t, 2H, benzimidazole-C5,6-H), 7.61 (s, 1H, NH, D2O exchangeable), 7.87 (d, 2H, J=7.80 Hz, benzimidazole-C4,7-H), 8.25 (s, 1H, NH, D2O exchangeable); MS m/z (% rel. Int.): 321 (1.06%, M++1), 320 (0.89%, M+.), 133.05 (100%, C7H7N3). Anal. Calcd. for C9H,6ClN3O2 (223.62) C, 48.34, H, 2.70, N, 18.79. Found: C, 48.34, H, 2.70, N, 18.79.
General procedure for the synthesis of N1-(benzimidzol-2-yl)-N2-(substituted amine)oxalamides 7(a,b)
A mixture of 6 (0.50 g, 0.22 mmol), either 4-amino-1,2-dihyodr-1,5-dimethyl-2-phenylpyrazol-3-one (0.45 g, 0.22 mmol) or 2-aminoanthracene- 9,10-dione (0.48 g, 0.22 mmol) and anhydrous potassium carbonate (0.30 g, 0.22 mmol) in mixed solvents of few drops of dry N,N-dimethylformamide and dry chloroform (20 ml) was heated under reflux for 5 h. After cooling, ice cold water (5 ml) was added with stirring. The separated solid was filtered, washed with water (2 x 20 ml), dried and crystallized from ethanol.
N1-(1H-benzimidzol-2-yl)-N2-(2,3-dihyodr-1,5-dimethyl-3-oxo-2-phenyl-1H-pyrazol-4-yl)oxalamide (7a)
Yield: 45.50%; M. P. 292-294°C; IR (KBr) ν max: 3420-3320, 3086, 2980, 1690-1651, 1562 cm-1; 1H-NMR (DMSO-d6, 400 MHz) δ (ppm)=1.72 (s, 3H, CH3), 2.74 (s, 3H, N-CH3), 6.9 (d, 2H, J=7.60 Hz, phenyl-C2,6-H), 7.21 (s, 1H, NH, D2O exchangeable), 7.24 (t, 1H, phenyl- C4-H), 7.37 (t, 2H, phenyl-C3,5-H), 7.42 (t, 2H, benzimidazole-C5,6-H), 7.5 (d, 2H, J=8.00 Hz, benzimidazole-C4,7-H), 7.53 (s, 1H, NH, D2O exchangeable), 8.67 (s, 1H, NH, D2O exchangeable); MS m/z (% rel. Int.): 391.20 (1.07%, M++1), 390.25 (0.54%, M+.), 368.20 (100%, C18H20N6O3). Anal. Calcd. for C20H15N7O2 (385.38): C, 61.53, H, 4.65, N, 21.53. Found: C, 61.71, H, 4.62, N, 21.72.
N1-(1H-benzimidzol-2-yl)-N2-(9,10-Dihyodr-9,10-dioxanthracen-2-yl)oxalamide (7b)
Yield 47.50%; M. P. 210-212°C; IR (KBr) ν max: 3433, 3380-3280, 3060, 1700, 1680, 1672, 1660, 1627, 1587 cm-1; 1H-NMR (DMSO-d6, 300 MHz) δ (ppm)=6.65 (s, 1H, NH, D2O exchangeable), 6.95 (d, 1H, J=8.40 Hz, anthraquinone-C3-H), 7.28 (s, 1H, NH, D2O exchangeable), 7.30 (s, 1H, anthraquinone-C1-H), 7.80-7.89 (t, 4H, anthraquinone-C6.7-H, benzimidazole-C5,6-H), 7.91 (d, 1H, J=8.10 Hz, anthraquinone-C4-H), 8.05 (s, 1H, NH, D2O exchangeable), 8.18 (d, 4H, J=6.00 Hz, anthraquinone-C5,8-H, benzimidazole-C4,7-H); 13C-NMR(DMSO-d6, 300 MHz): 109.64 (benzimidazole-C4,7), 118.05 (anthraquinone-C1), 121.13 (benzimidazole-C5,6), 126.25 (anthraquinone-C3), 126.30 (anthraquinone-C4,5,8), 128.193 (anthraquinone-C6,7,5a,8a), 129.47 (anthraquinone-C4a), 132.98 (anthraquinone-C2), 133.22 (benzimidazole1a,3a, anthraquinone-C1a), 134.20 (benzimidazole-C2), 180.03(2 acetyl-C=O), 183.29 (anthraquinone-C9,10). Anal. Calcd. for C23H14N4O4 (410.38): C, 67.31, H, 3.44, N, 13.65. Found: C, 67.44, H, 3.41, N, 13.73.
2-(9,10-Dihyodr-9,10-dioxoanthracen-2-ylamino)-N-(1H-benzimidazol-2-yl)acetamide (9)
A mixture of 8 (0.46 g, 0.22 mmol), 2-aminoanthracene-9,10-dione (0.48 g, 0.22 mmol) and anhydrous potassium carbonate (0.30 g, 0.22 mmol) in in mixed solvents of few drops of dry N,N-dimethylformamide and dry chloroform (20 ml) was heated under reflux for 5 h. After cooling, ice cold water (5 ml) was added with stirring. The separated solid was filtered, washed with water (2 x 20 ml), dried and crystallized from ethanol.
Yield 42 %; M. P. 218-220°C; IR (KBr) ν max: 3433, 3346, 3221, 3064, 2954, 1690, 1674, 1660, 1627, 1587 cm-1; 1H-NMR (DMSO-d6, 300 MHz) δ (ppm)=3.12 (s, 2H, CH2-H), 6.65 (s, 1H, NH, D2O exchangeable), 6.95 (d, 1H, J=8.40 Hz, anthraquinone-C3-H), 7.29 (s, 1H, NH, D2O exchangeable), 7.30 (s, 1H, anthraquinone-C1-H), 7.80 -7.89 (t, 4H, anthraquinone-C6,7-H, benzimidazole-C5,6-H), 7.91 (d, 1H, J=8.10 Hz, anthraquinone-C4-H), 8.15 (d, 4H, J=6.00 Hz, anthraquinone-C5,8-H, benzimidazole-C4,7-H), 8.19 (s, 1H, NH, D2O exchangeable); 13C-NMR( DMSO-d6, 300 MHz): 78.9 (CH2), 109.5 (anthraquinone-C3), 110 (anthraquinone-C1), 118.2 (anthraquinone-C4a), 121.07 (benzimidazole- C5,6), 122 (anthraquinone-C6,7,5a,8a), 126.48 (anthraquinone-C4,5,8), 129.64 (benzimidazole-C4,7), 133.26 (anthraquinone-C2), 134.2 (benzimidazole1a,3a, anthraquinone-C1a), 151.1 (benzimidazole-C2), 180.7 (acetyl-C=O), 184 (anthraquinone-C9,10); MS m/z (% rel. Int.): 398 (0.62%, M++2), 397 (0.20%, M++1), 396 (0.11%, M+.), 223 (100%, C14H8NO2). Anal. Calcd. for C23H16N4O3 (396.4): C, 69.69, H, 4.07, N, 14.13. Found: C, 69.83, H, 4.11, N, 14.19.
Cytotoxic activity studies
Cytotoxic activity studies were conducted at Ain Shams University, Faculty of Pharmacy, Pharmacology Department and national cancer institute, Cairo University. Antioxidants status assays were conducted at National Research Centre, Biochemistry Department, Division of Genetic Engineering and Biotechnology.
All compounds were tested using Sulfo-Rhodamine-B (SRB) assay for cytotoxic activity against Breast carcinoma cell line (MCF-7) human lung carcinoma A549 and human epitheloid cervix carcinoma HELA at a dose of (100 ug/ml).
Measurement of potential cytoyoxicity by SRB assay against cisplatin
Cytoyoxicity was determined using SRB [22] and the compounds were tested against cisplatin. Exponentially growing cells were collected using 0.25% Trypsin-EDTA and seeded in 96-well plates at (104 cells/ well) in RPMI-1640 supplemented medium. After 24 h, cells were incubated for 72 h with various concentrations of the tested compounds. Following 72 h treatment, the cells will be fixed with 10% trichloroacetic acid for 1 h at 4ºC. Wells were stained for 10 min at room temperature with 0.4% SRB dissolved in 1% acetic acid. The plates were air dried for 24 h and the dye was solubilized with Tris-HCl for 5 min on a shaker at 1600 rpm. The optical density (OD) of each well was measured spectrophotometrically at 564 nm with an ELISA microplate reader (ChroMate-4300, FL, USA). The IC50 values were calculated according to the equation for Boltzman sigmoidal concentration–response curve using the nonlinear regression fitting models.
Antioxidants status assays
Antioxidant enzyme assays
The cells in culture medium were treated with 20 μl of compound 5a and then incubated for 24 h at 37°C, in a humidified 5% CO2 atmosphere. The MCF-7, HELA and A549 cells were harvested and homogenates were prepared in saline using a tight pestle homogenizer until complete cell disruption for further biochemical analysis. The supernatant obtained after centrifugation of cell homogenates at 3000 g was used for determination of activities of enzymes including superoxide dismutase [23], catalase [24], glutathione peroxidase [25].
Oxidative stress assays
The levels of hydrogen peroxide (H2O2) [26], nitric oxide [27] (NO) and reduced glutathione (GSH) [28] were determined.
Estimation of nucleic acids and protein
Nucleic acids (DNA and RNA) and total protein were precipitated and measured in cell homogenates. Total DNA was extracted and assayed [29]. Total RNA was extracted and assayed according to the method adopted from the method provided by Hybaid/AGS (Germany), and total cellular protein was assayed by the method of Lowry and associates [30].
Statistical analysis
The results are reported as Mean ± Standard error (S. E.) for at least four times. Statistical differences were analyzed according to one way ANOVA test followed by student's t test wherein the differences were considered to be significant at p<0.05.
Results and Discussion
Chemistry
The synthesis of new benzimidazole derivatives was achieved through schemes 1 and 2. The structure of the newly synthesized compounds was confirmed by elemental analysis and spectral data (IR, 1H-NMR, 13C-NMR and MS).
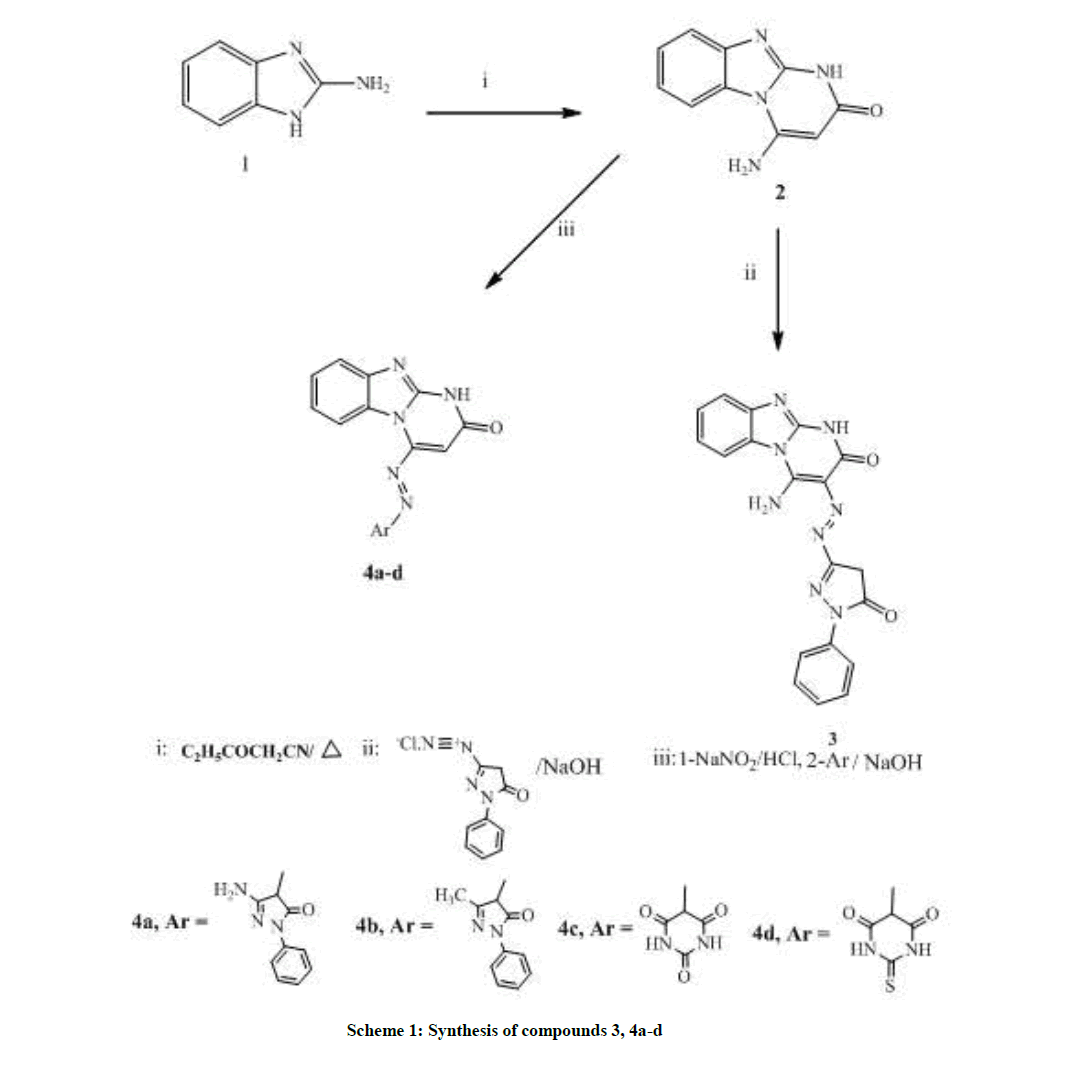
Scheme 1: Synthesis of compounds 3, 4a-d
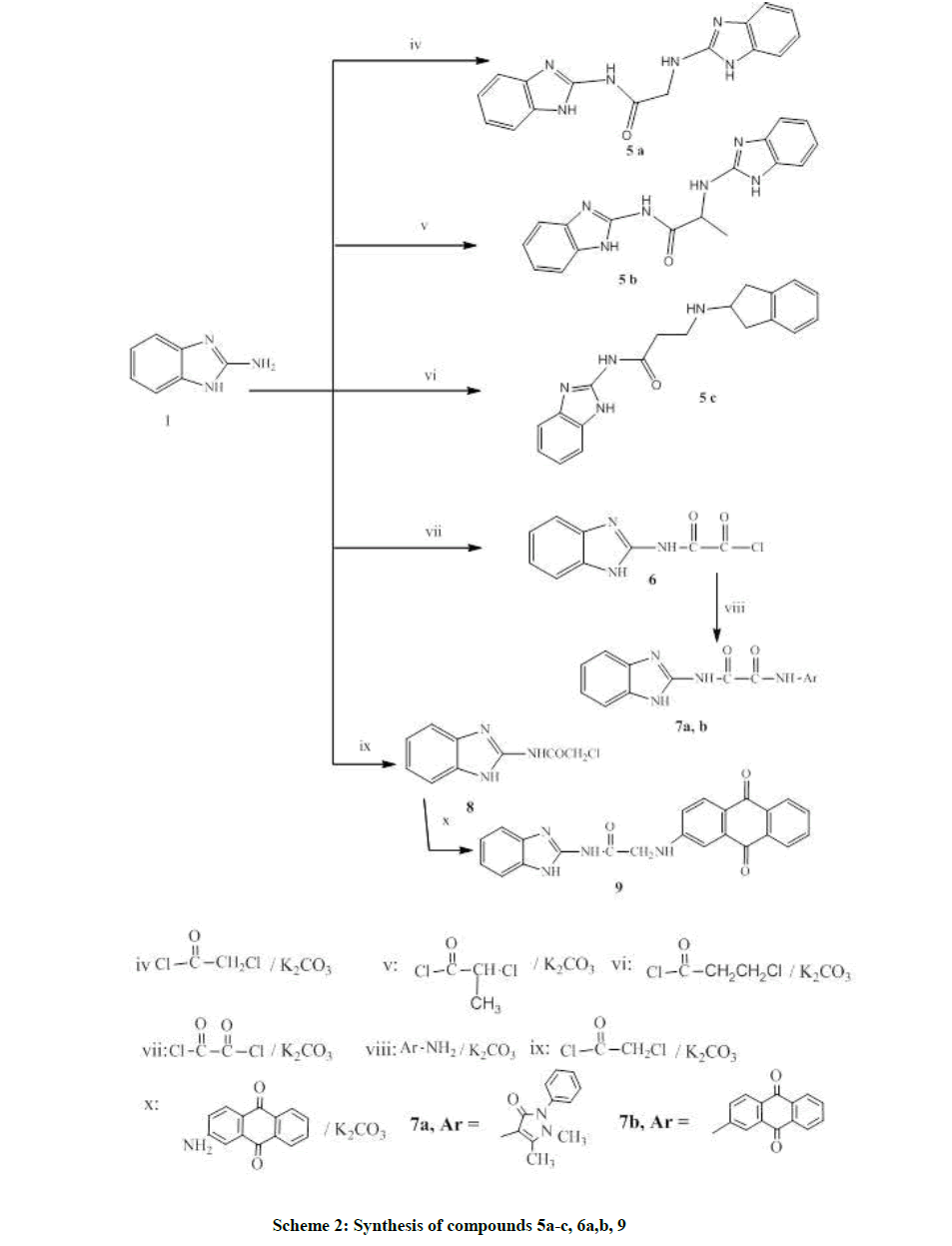
Scheme 2: Synthesis of compounds 5a-c, 6a,b, 9
Based on incorporation of benzimidazole scaffold at N1 position of the imidazole ring, scheme 1 utilizes the synthesis of different azo coupling derivatives, starting with 4-amino-1H-benzo[4,5]imidazo[1,2-a]pyrimidin-2-one (2) which was prepared via heating 2-aminobenzimidazole (1) with excess ethyl cyanoacetate at 150°C [16]. Then 2 was conjugated with either pyrazolo or pyrimido moieties by a coupling reaction at position 4 & 5 producing compounds 3 & 4 due to the great potential of both moieties as anti-cancer agents.
Synthesis of compound 3 [31] was achieved by reaction of active methylene of compound 2 with diazotized 3-amino-1-phenyl-1H-pyrazol- 5(4H)-one. The structure of 3 was proven by the appearance of 2 CO bands at 1675, 1670 cm-1. In addition, 1H-NMRspectrum revealed the disappearance of singlet signal equivalent to pyrimidinone-C5-H at range 7.5-8.5 ppm and appearance of singlet signal of pyrazole-C4-H at δ 4.99 ppm.
Preparation of 4a-d was attained following a reported procedure for the preparation of analogous compounds [10] by reacting compound 2 with sodium nitrite in acid medium producing the diazonium salt which was added to a basic solution of various compounds with active methylene group in sodium hydroxide solution. 2 and 3 bands equivalent to 2 & 3 C=O groups were distinctive of the produced derivatives in addition, the added peaks in 1H-NMR, where 4a was detected by the singlet signal at δ 6.18 ppm equivalent to the 2H of the NH2 group. On the other hand 4b structure was proven by the appearance of singlet signals at δ 2.07 of the methyl group. Both 4c & 4d structures were confirmed by having 3 & 4 CO bands in IR and their 1H-NMR spectra showing singlet signal at 4, 99 & 5.03 of barbituric & thiobarbituric-C5-H.
Scheme 2 is concerned with dimer synthesis of new derivatives incorporating variable spacers and different polarity which were designed in the synthesis of compounds 5a-c by one pot procedure. The reaction could be performed in presence of catalyst as anhydrous potassium carbonate or tri-ethylamine [15].
Additionally, scheme 2 utilizes the usage of anthraquinone moiety mimicking both Doxorubicin and Daunorubicin as antitumor agents of anthracycline® class in the synthesis of compounds 7a, b and 9. The synthesis firstly involves the formation of precursor 5a-c by one pot procedure. The usage of amines to acid halides in a ratio 2: 1 was crucial to achieve the desired product.
Compound 6 was reported without separation or isolation and hence, no physical or spectral data was available for identification [17-19]. In this paper it was synthesized by refluxing 2-aminobenzimidazole with oxalyl dichloride in the presence of anhydrous potassium carbonate in dry benzene to allow simple separation of the product.
Furthermore, compounds 7a, b were synthesized by refluxing compound 6 with two amines (4-aminoantipyrane and 2-aminoanthracene-9,10- dione) using anhydrous potassium carbonate as catalyst. N,N-dimethylformamide (few drops) was added to increase the reactants solubility and the product yield.
Compound 8 was prepared by refluxing 2-aminobenzimidazole and chloroacetyl chloride in THF [18], dry benzene [20] or dry acetone [21]. Following the reported procedure [20], compound 8 was synthesized in dry benzene. Compound 9 was produced by refluxing a mixture of 8 and 2-aminoanthracene-9,10-dione using anhydrous potassium carbonate in dry benzene.
Cytotoxic effect
All the newly synthesized compounds were tested for their cytotoxic activity against three different cell lines namely human breast cancer cell line (MCF-7), human lung carcinoma (A549) and human epitheloid cervix carcinoma (HELA) using Sulforhodamine B (SRB) colorimetric assay, comparing to cisplatin as reference drug. The results were represented in (Table 1) as percentage inhibition values. It was found that compounds 5a and 7b revealed highest activity on three cell lines, where compounds 4a, 4b, 4d and 9 possess intermediate antitumor activities (Figure 2).
| Compound | MCF-7 | HELA | A549 |
|---|---|---|---|
| Cisplatin | 60 | 35 | 60 |
| 3 | 46 | 36 | 70 |
| 4a | 58 | 61 | 77 |
| 4b | 40 | 54 | 77 |
| 4c | 44 | 65 | 75 |
| 4d | 30 | 60 | 72 |
| 5a | 95 | 54 | 77 |
| 5b | 40 | 54 | 73 |
| 5c | 40 | 35 | 75 |
| 7a | 45 | 32 | 67 |
| 7b | 80 | 35 | 72 |
| 9 | 62 | 37 | 61 |
Table 1: Percentage inhibition of tested compounds on three cell lines: breast cancer MCF-7, human lung carcinoma A549 and human epitheloid cervix carcinoma HELA at a dose of (100 ug/ml)
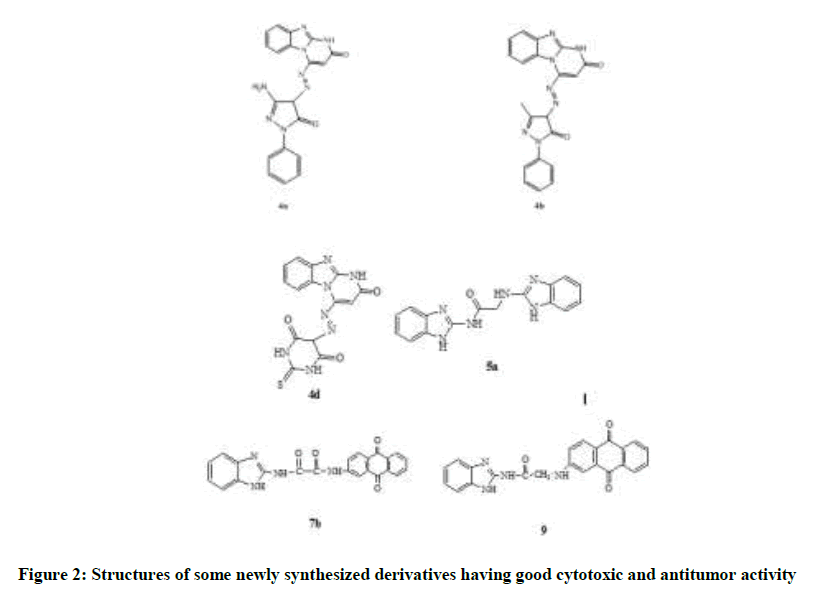
Figure 2: Structures of some newly synthesized derivatives having good cytotoxic and antitumor activity
Some structural features that are important for explanation of their cytotoxic effects showed that compound 7b was better in activity than both 7a and 9 and this may be due to the extra hydrogen bonding. Furthermore, the acetyl linker of compound 5a was the most preferable one in comparison with the prop-2-yl and the prop-3-yl chains of both 5b and 5c. Moderate effect was achieved by compound 4a and was better than 4b may be attributed to the hydrogen bonding ability.
From the previous, compounds 7b, 5a, 9 and 4a showed the highest activity amongst all derivatives. Both 7b and 9 shared the same tricyclic anthraquinone moiety which contributes to the better activity; following was 5a with the dimer structure having acetyl linker showing better activity than prop-2-yl and the prop-3-yl linkers. On the other hand, 4a of smaller structure but its activity may be attributed to the added hydrogen bond formation with the receptor.
Moreover, 4d, 4b, 7a, 5c and 5b showed moderate activity compared to other derivatives and to cisplatin and this may be due to the hydrazine derivatives even when bulky molecules where used showed lesser activity comparable to other tricyclic or dimer analogues, also, the longer and branched linkers of 5b, c may be the cause of its moderate activity.
Antioxidant effect
The most potent cytotoxic derivative 5a was submitted to further assays to investigate the mechanism by which it exerts its anticancer activities. We estimated the activities of the free-radical-metabolizing enzymes, including superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GSH-Px), the levels of the oxidative stress parameters including hydrogen peroxide (H2O2), nitric oxide (NO) and reduced glutathione (GSH) in MCF-7 cells treated with the prepared compounds and the effect of these compounds on the levels of total protein and nucleic acids.
As shown in (Table 2), general treatment of the cells with different compounds and cisplatin resulted in a significant increase in the activity of SOD and a level of H2O2 higher than those of control, accompanied with a significant depletion in the activity of CAT, and GSH-Px, and the level of GSH These changes were in the order of 5a more than cisplatin, which is in the accordance with the order of cytotoxicity activity of the tested compound, indicating an increase in the cellular levels of reactive oxygen species. These results stated that the antitumor effect of the present compound might be exerted at least partly by production of reactive oxygen species. As shown in (Table 3), the level of total protein and nucleic acids were significantly lower than of control, while the level of NO was significantly higher in MCF-7 cells treated with the compound, compared to control cells. The activity of compound 5a resulted in high SOD activity and H2O2 and low activity of CAT and GSH-Px as well as GSH level. The antitumor activity of compound 5a was accompanied by increases in SOD activity of tumor-treated cells compared to control cells. This means that this compound can increase H2O2 production. The H2O2 produced should be rapidly removed through the activation of both CAT and GSH-Px enzymes. The present results show that activities of CAT and GSH-Px and the level of reduced GSH were lowered in groups treated with compounds compared to control cells. Consequently, the excess H2O2 produced in tumor cells with the compound cannot be removed. In other words, the accumulation of H2O2 and other free radicals in tumor cells should be partly the cause of tumor cell killing. Thus, the results of the present study were consistent with the hypothesis that the prepared compound exerts their antitumor effects because they produce reactive oxygen species (ROS).
| Treatment (µg / ml) |
SOD U/mg Protein |
CAT U/mg Protein |
GSH-Px U/mg Protein |
GSH nmol/mg protein |
H2O2 nmol/mg protein |
|---|---|---|---|---|---|
| Control (DMSO) | 43.30 ± 4.70 | 8.30 ± 0.88 | 10.00 ± 1.20 | 46.30 ± 5.00 | 18.90 ± 2.10 |
| Cisplatin | 122.80 ± 14.45a | 3.66 ± 0.37a | 5.37 ± 0.60a | 25.00 ± 2.85a | 50.20 ± 5.50a |
| 5a | 135.44 ± 11.70a,b | 3.11 ± 0.30a | 4.95 ± 0.55a | 21.96 ± 3.00a | 54.30 ± 6.40a |
Data are expressed as means ± S. E. of four separate experiments; a and b is significant difference from control and cisplatin groups respectively at (p < 0.05)
Table 2: Effect of treatment with the prepared compounds on the activities of superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GSH-Px), as well as the levels of reduced glutathione (GSH) and hydrogen peroxide (H2O2) in MCF-7 treated cells
| Compounds | Protein (µg/106 cells) |
RNA (µg/106 cells) |
DNA (µg/106 cells) |
NO (µmol/mg protein) |
|---|---|---|---|---|
| Control (DMSO) | 145.00 ± 15.30 | 18.60 ± 1.90 | 9.80 ± 1.10 | 2.20 ± 0.24 |
| Cisplatin | 65.70 ± 7.50a | 4.60 ± 0.50a | 4.80 ± 0.54a | 4.60 ± 0.50a |
| 5a | 50.20 ± 6.50a,b | 3.90 ± 0.46a | 4.80 ± 0.53a,b | 5.00 ± 0.60a |
The values are expressed as mean ± SE of four separate experiments; a and b is significant difference from control and cisplatin groups respectively at (p < 0.05)
Table 3: Effect of prepared compounds on the level of total protein, nucleic acids (RNA and DNA) and nitric oxide (NO) in MCF-7 treated cells
Conclusion
In conclusion, the present results suggest that the newly synthesized compounds possess good to moderate cytotoxic effect. Compounds 5a, 7b revealed significant cytotoxic activity comparable to the activity of commonly used anticancer drug, cisplatin. They exert their effects by regulating free radicals production by increasing the activity of superoxide dismutase and depleting intracellular reduced glutathione, catalase, glutathione peroxidase, accompanied with high production of hydrogen peroxide, nitric oxide and other free radicals causing tumor cells death, as monitored by reduction in the synthesis of protein and nucleic acids. The results revealed that these compounds might act as potent cytotoxic agent in the future clinical trials.
Acknowledgement
The authors would like to express their gratitude to the dean of Faculty of Pharmacy Cairo University, Cairo, Egypt.
References
- P.P. Prabhu, T. Panneerselvam, C.S. Shastry, A. Sivakumar, S.S. Pande, J. Saud. Chem. Soc., 2015, 19, 181-185.
- M.S. Bashandy, M.S. ElSaid, R.K. Arafa, M.M. Ghorab, J. Enzym. Inhib. Med. Chem., 2014, 29, 619-627.
- R.A. Smith, V. Cokkinides, O.W. Brawley, C. A. Cancer J. Clin., 2009, 59, 27-41.
- M.M. Ghorab, M.S. Alsaid, M. Higgins, A.T. Dinkova-Kostova, A.A. Shahat, N.H. Elghazawy, R.K. Arafa, J. Enzyme Inhib. Med. Chem., 2016, 31, 1612-1618.
- Y. Yan, Z. Liu, J. Zhang, R. Xu, X. Hu, G. Liu, Bioorg. Med. Chem. Lett., 2011, 21, 4189-4192.
- Y. Bansal, O. Silakari, Bioorg. Med. Chem., 2012, 20, 6208-6236.
- P. Biju, K. McCormick, R. Aslanian, M. Berlin, D. Solomon, H. Wang, Y.J. Lee, R. Bitar, D. Prelusky, R. Mcleod, Y. Jia, X. Fernandez, S. Eckel, A. House, G. Lieber, J. Jimenez, G. Kelly, R. Chapman, J. Phillips, J. Anthes, Bioorg. Med. Chem. Lett., 2012, 22, 1086-1090.
- B. Mathew, J. Suresh, S. Anbazhagan, J. Saud. Chem. Soc., 2016, 20, 132-139.
- C.G. Neochoritis, T. Zarganes-Tzitzikas, C.A. Tsoleridis, J. Stephanidou-Stephanatou, C.A. Kontogiorgis, D.J. Hadjipavlou-Litina, T. Choli-Papadopoulou, Eur. J. Med. Chem., 2011, 46, 297-306.
- A.T. Taher, N.A. Khalil, E.M. Ahmed, Y.M. Ragab, Chem. Pharm. Bull., 2012, 60, 778-784.
- M. Shaharyar, M.M. Abdullah, M.A. Bakht, J. Majeed, Eur. J. Med. Chem.,2010, 45, 114-119.
- T.S. Reddy, H. Kulhari, V.G. Reddy, V. Bansal, A. Kamal, R. Shukla, Eur. J. Med. Chem., 2015, 101, 790-805.
- Y. Kubota, H. Fujii, J. Liu, S. Tani, Nucleic Acids Res. Suppl.,2001,1, 101-102.
- S. Alper, Ö.T. Arpaci, E.Ş. Aki, I. Yalçin, IL Farmaco., 2003, 58, 497-507.
- A.T. Taher, G.H. Hegazy, Arch. Pharm. Res., 2013, 36, 573-578.
- F. Karcı, A. Demirçalı, İ. Şener, T. Tilki, Dyes Pigm., 2006, 71, 90-96.
- G. Primofiore, A.M. Marini, F.D. Settimo, J.S. Franzone, U. Mason, M.C. Reboani, R. Cirillo, Eur. J. Med. Chem., 1988, 23, 397-401.
- P. Caroti, C. Ceccotti, F.D. Settimo, G. Primofiore, IL Farmaco., 1989, 44, 227-255.
- A. D. Settimo, A. M. Marini, G. Primofiore, F. D. Settimo, IL Farmaco., 1995, 50, 321-326.
- A. Mobinikhaledi, M. Kalhor, M. Hatami, Heterocycl. Commun.,2010, 16, 165-170.
- A. Mobinikhaledi, N. Foroughifar, M. Kalhor, M. Mirabolfathy, J. Heterocyclic Chem., 2010, 47, 77-80.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J.T. Warren, H. Bokesch, S. Kenney, M.R. Boyd, J. Natl. Cancer Inst., 1990, 82: 1107-1112.
- D.E. Paglia, W.N. Valentine, J. Lab. Clin. Med., 1967, 70, 158-169.
- H. Aebi, New York: Academic press., 1984, 673-679.
- S. Marklund, G. Marklund, Eur. J. Biochem.,1974, 47, 469-474.
- S.P. Wolff, Methods Enzymol., 1994, 233, 182-189.
- H.A.C. Montgomery, J.F. Dymock, Analyst., 86, 414-417.
- G.L. Ellman, Arch. Biochem. Biophys., 1959, 82, 70-77.
- T. Zhou, G. Zhou, W. Song, N. Eguchi, W. Lu, E. Lundin, T.G. Nordberg, Toxicology., 1999, 142, 1-13.
- O.H. Lowry, N.J. Rosebrough, A.L. Farr, R.J. Randall, J. Biol. Chem., 1951, 103, 265-275.
- M. Shabaan, A.T. Taher, E.O. Osman, Eur. J. Med. Chem., 2011, 2, 365‐371.



