Research Article - Der Pharma Chemica ( 2021) Volume 13, Issue 2
Quantitative Structure-Activity Relationship and Molecular Modeling Studies on a Series of Hydroxypyrrolo[2,1-c][1,4]Benzodiazepine-5,11-dione Acting as Angiotensin Converting Enzyme I Inhibitors
Ayesha Sanober and Neeraj Agarwal*Neeraj Agarwal, Department of Biotechnology, Meerut Institute of Engineering and Technology, Meerut 250005, India, Email: neeraj.agarwal@miet.ac.in
Received: 15-Sep-2020 Accepted Date: Feb 20, 2021 ; Published: 26-Feb-2021
Abstract
Angiotensin-Converting Enzyme (ACE) 1 shows myriad activities that can be associated with the rennin-angiotensin system. From hypertension and electrolyte balance to oxidative stress, ACE contributes to various functions in the body. The versatility of these inhibitors makes them an interesting subject to ponder upon. However, the purpose of this paper is to study a series of ACE inhibitors and predict yet better compounds of the series. Hence a series of hydroxypyrrolo[2,1-c][1,4]benzodiazepine-5,11-dione compounds have been taken, and a quantitative structure-activity relationship (QSAR) study followed by modeling of molecules has been performed upon them, Obtained a cross-validated result (r2 cv) of 0.713 carried out by Leave one out method (LOO), and predicted (r2 press)=0.716 with the coefficient of correlation of (r) obtained by the multiple regression analysis is 0.944. A new series of compounds have been thus proposed based upon the QSAR model generated. These compounds were docked with the protein and ADME properties of each of the newly proposed compounds were studied using Swiss ADME that highlights physiochemical properties of the compounds, their lipophilicity, solubility, drug likeliness, pharmacokinetics, etc. Toxicity prediction of these compounds is also made, making them suitable leads against ACE 1.
Keywords
Angiotensin-Converting Enzyme (ACE), Molecular Docking, QSAR, ADME, Toxicity, Drug likeness
Introduction
ACE (angiotensin I converting enzyme 1) act as a key component of the renin-angiotensin system. It involves formation of angiotensin II from angiotensin I along with the degradation of bradykinin. As a part of renin-angiotensin system, it was found that ACE plays a key role in homeostatic mechanism of mammals, thus regulates blood pressure and electrolytic balance in the body [1]. Hypertension can be controlled by the regulating the ACE activity along with that, ACE also plays an important role in the therapies for heart failure and Diabetes nephropathy[2]. They change the balance between the salt-retentive, vasoconstrictive, and hypertrophic properties of angiotensin II and the vasodilatory and natriuretic properties of bradykinin [3]. In addition to the aforementioned activities, emerging evidence suggests that ACE inhibitors have important implications for vascular oxidative stress [4]. These inhibitors are different from each other in terms of the chemical structure of their active components, in their potency, in bioavailability, in plasma half-life, in their route of elimination, in distribution and their affinity for tissue-bound ACE, and in whether or not they are administered as prodrugs [5]. The versatility of these inhibitors makes them an interesting subject to ponder upon. However, the purpose of this paper is to study a series of ACE inhibitors and predict yet better compounds. A number of ACE inhibitors are already being used to combat high blood pressure and heart ailments, some of which are Benazepril, Captopril and Lisinopril.
The process of drug discovery has always been an extremely time consuming, labour intensive, and expensive process. A more cost-effective solution to this problem is rational designing of drugs using in-silico methods via molecular modelling, simulation, and virtual screening for the identification of promising candidates even before their chemical synthesis [6]. Quantitative structure-activity relationship (QSAR) modelling is one of the most widely used computer-aided tools used in medicinal chemistry for drug discovery and lead optimization [7]. In a previous study, Dinesh Addla and co-workers have reported 10-substituted 2-hydroxypyrrolobenzodiazepine-5,11-dione analogues as potent Angiotensin converting enzyme (ACE) inhibitors [8]. In the current study we have created a QSAR model to introduce some new compounds of this series that are expected to show better activity and lesser toxicity. The ADME properties and toxicity of these compounds have also been studied and evaluated.
Materials and Methods
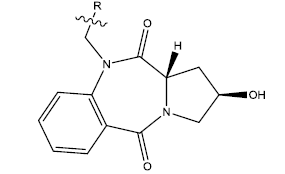
We have taken a series of hydroxypyrrolo[2,1-c][1,4]benzodiazepine-5,11-dione (5a–w) compounds. All the compounds of this series are listed in Table 1 along with some parameters that were found to regulate their activities. The most significant parameters were found to be PPSA (Atomic charge weighted positive surface area), MATSV (Moran autocorrelation-lag3/weighted by atomic van der Waals volumes), TD (Topological Diameter) and NALC (Number of atoms in the largest chain). A QSAR model was thus designed based upon these parameters. All of these parameters were calculated using Chem Des, which is a free web-based platform for the calculation of molecular descriptors and fingerprints. Each of the significant parameters is found to have negligible mutual correlation (Table 2). 10 new compounds of the series were also designed using Swiss ADME. Molecular docking has been performed to study the interactions between the protein taken from Protein Databank (PDB ID 1O86) and the proposed compounds (Table 3) and also with the licensed compounds. ADME properties were checked to see if these compounds can serve as a potent drug. Water solubility, pharmacokinetics, drug likeliness, Medicinal chemistry and Lipophilicity were checked using 'swiss ADME'. Further, the toxicity of each of these compounds was checked using 'Pro Tox'. This web server has an easy-to-use interface and it only requires a 2D structure of the molecule whose toxicity has to be predicted [9]. Hepatotoxicity, carcinogenicity, cytotoxicity, mutagenicity and immunotoxicity of each of the proposed compounds were specifically checked.
 |
||||||||
| S.no | R | PPSA | MATSV | NALC | T D | log(1/IC50) | ||
| obsda | Calculated | Pred (LOO) | ||||||
| 1. | H | 24.186 | -0.045 | 37 | 10 | 6.04 | 6.10 | 6.13 |
| 2. | 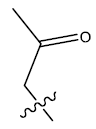 |
23.801 | 0.012 | 32 | 10 | 6.03 | 6.02 | 6.13 |
| 3. | 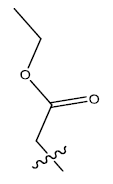 |
32.751 | 0.057 | 41 | 12 | 6.05 | 6.08 | 5.96 |
| 4. | 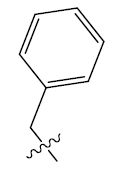 |
25.874 | -0.014 | 42 | 12 | 6.15 | 6.09 | 6.11 |
| 5c. | 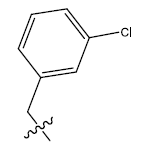 |
30.42 | -0.001 | 42 | 12 | 5.92 | 6.17 | - |
| 6b. | 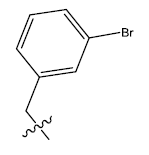 |
29.44 | 0.044 | 42 | 12 | 6.24 | 6.11 | - |
| 7. | 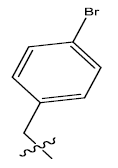 |
25.502 | 0.044 | 42 | 12 | 6.17 | 6.16 | 6.17 |
| 8c. | 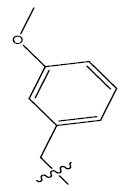 |
30.06 | -0.049 | 46 | 13 | 5.90 | 6.18 | - |
| 9b. | 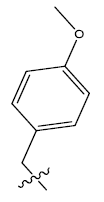 |
31.09 | -0.049 | 46 | 14 | 6.05 | 5.95 | - |
| 10. | 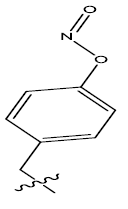 |
26.433 | -0.111 | 44 | 13 | 5.95 | 5.96 | 5.97 |
| 11. | 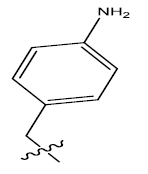 |
29.894 | -0.045 | 44 | 13 | 6.01 | 6.00 | 6.01 |
| 12. | 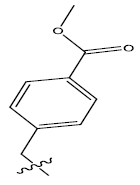 |
48.852 | -0.097 | 48 | 15 | 5.7 | 5.72 | 5.76 |
| 13. |  |
36.119 | -0.082 | 45 | 13 | 5.98 | 5.93 | 5.94 |
| 14b. |  |
26.97 | -0.082 | 42 | 12 | 6.09 | 6.00 | - |
| 15. |  |
32.109 | -0.106 | 44 | 13 | 5.96 | 5.91 | 5.92 |
| 16. |  |
28.287 | -0.101 | 42 | 12 | 6.03 | 5.98 | 5.99 |
| 17. |  |
31.809 | -0.103 | 42 | 12 | 5.91 | 5.94 | 5.94 |
| 18. |  |
37.53 | 0.039 | 54 | 14 | 6.25 | 6.21 | 6.15 |
| 19. |  |
28.186 | -0.111 | 42 | 12 | 5.95 | 5.97 | 6.00 |
| 20. |  |
28.924 | 0.024 | 44 | 13 | 6.03 | 6.08 | 6.09 |
| 21b. |  |
28.23 | 0.058 | 44 | 13 | 6.07 | 6.11 | - |
| 22. |  |
24.189 | 0.048 | 39 | 11 | 6.18 | 6.17 | 6.16 |
| 23. |  |
32.221 | -0.055 | 51 | 14 | 6 | 6.08 | 6.12 |
Table 1: 2-hydroxypyrrolobenzodiazepine-5, 11-dione analogues, their physiochemical parameters and ACE inhibition activity.
| S.no | Predicted Compounds | PPSA | MATSV | NALC | T D | log(1/IC50) |
|---|---|---|---|---|---|---|
| 1. |  |
25.735 | -0.038 | 44 | 10 | 6.27 |
| 2. | 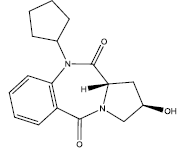 |
21.526 | -0.038 | 42 | 10 | 6.25 |
| 3. | 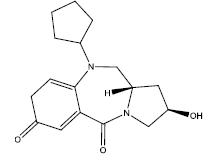 |
21.348 | -0.077 | 44 | 10 | 6.27 |
| 4. | 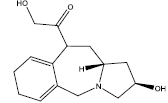 |
24.60 | 0.216 | 40 | 10 | 6.44 |
| 5. | 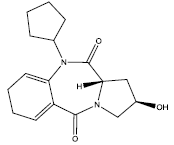 |
21.342 | -0.038 | 44 | 10 | 6.31 |
| 6. | 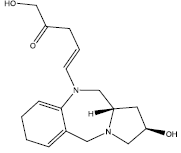 |
35.025 | 0.206 | 46 | 13 | 6.26 |
| 7. | 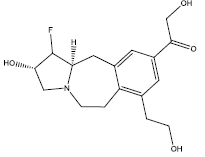 |
32.45 | 0.269 | 45 | 12 | 6.40 |
| 8 | 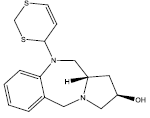 |
23.077 | 0.2 | 41 | 10 | 6.50 |
| 9. | 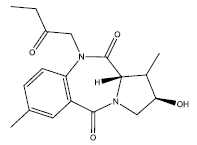 |
23.72 | -0.015 | 46 | 11 | 6.29 |
| 10. | 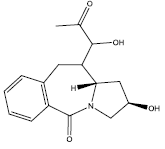 |
24.41 | 0.03 | 40 | 10 | 6.30 |
Table 2: Some proposed compounds belonging to the series of Table 1 and their activities predicted from (1).
| PPSA | MATSV | NALC | TD | |
|---|---|---|---|---|
| PPSA-3 | 1.000 | 0.017 | 0.220 | -0.59 |
| MATSv3 | 1.000 | -0.02 | 0.250 | |
| nAtomLC | 1.000 | -0.68 | ||
| Topological dia | 1.000 |
Table 3: Correlation between the significant parameters
Results and Discussions
QSAR results
All the compounds present in Table 1 have been divided into two sub-Categories: a training set and test set. The selection of test set compounds was done randomly by keeping in mind that, there is a significant variation in their structures and activity. All the test set compounds are denoted by superscript "b" and the outliers are denoted by a superscript "c" in Table 1. All the other compounds were grouped under the training set. A multiple regression analysis (Hansch analysis) performed on the compounds of the training set, revealed the following correlation.
log(1/IC50)= -0.009(± 0.007)PPSA + 1.047(± 0.436) MATSV + 0.027(± 0.013) NALC - 0.079(± 0.062) TD + 6.143(± 0.275) (1)
n = 17, r = 0.944, s = 0.048, F (4,12) = 24.417 (5.41)
r2 cv= 0.713, r2pred = 0.716
The most significant parameters were found to be PPSA (Atomic charge weighted positive surface area), MATSV (Moran autocorrelationlag3/ weighted by atomic van der Waals volumes), TD (Topological Diameter) and (Number of atoms in the largest chain). Table 3 suggests that there is no significant mutual correlation between the parameters of significance. IC50 refers to amount (molar concentration) of compound required for 50% inhibition of the enzyme. Among the statistical parameters in (1), n stands for the number of compounds present in the training set, r stands for correlation coefficient, r2 cv is defined by the square of the cross-validated correlation coefficient produced from leave-one-out (LOO) jackknife procedure, S stands for standard deviation, F stands for Fischer ratio between the variances of calculated and observed activities, and the data within the parentheses with ± sign are 95% confidence intervals. The figure within the parenthesis stands for the standard-value at the 99% level. The values of these statistical parameters show that the correlation obtained is quite significant.
The correlation expressed by (1) appears to be extremely significant, and r2 cv and r2 pred values can help judge the internal and external validation. The r2 cv is calculated by the formula
 (2)
(2)
where, yi,obsd and yi,pred is observed and predicted(from LOO) activity values of the compound, respectively, and yav,obsd is the average of observed activities of all compounds used in the correlation. The correlation turns out to be valid if r2 cv> 0.60. This indicates that the correlation expressed by (1) is indeed quite valid. However, the predictive capability of a model can most efficiently be judged by predicting the biological activity of the compounds belonging to the test set and the value of r2 pred that comes from the test set, which is defined as follows:
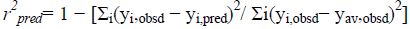 (3)
(3)
where, yi,obsd and yi,pred are referred to as observed and the predicted biological activity values of test set compounds and yi,obsd is same as it is in eqn(2). The values of biological activity of the test set compounds predicted from this equation are given in Table 1. Comparing the values we got to know that the predicted values are in fine agreement with the corresponding observed ones.
Docking Results
Molecular Docking is an optimization problem, where the aim is to find the ligand binding mode with the lowest potential energy. The process of Docking involves sampling the coordinate space of the target binding site and scoring each possible ligand pose within that site; the highest scoring poses then taken as the predicted binding mode for that compound. In order to perform docking studies and to check the interactions between the predicted compounds and the protein (PDB ID 1O86), Docking was performed using Molegro Virtual Docker whose results are given in Table 10.
The program MVD has four search algorithms and four native scoring functions. In this case 'plant score' scoring function was used along with 'iterated simplex' search algorithm that yielded an RMSD value of 1. The predicted compounds have hydrogen bond numbers comparable to commercial drugs. For all the compounds, all possible hydrogen bonds are shown in Table 4. It can also be seen that the overall energy of interaction with the enzyme for each predicted compound is comparable to the marketed compounds, and so is the case with the docking score of each predicted compound. All the compounds have a matching pose.
| Compound | Total Interaction energy | H-bond energy | No. of H- bonds | H- bonds | H- bond length(Å) | Mole dock score | Internal pose |
|---|---|---|---|---|---|---|---|
| 1 | -95.1334 | -2.8373 | 2 | O(8)-His(343 | 3.14 | -8.5912 | 7.54234 |
| O(8) - Asp(375) | 2.87 | ||||||
| 2 | -97.391 | -4.74556 | 2 | O(4) - Asp(409) | 3.08 2.86 | -91.1 | 6.229061 |
| O(8) - Gln(241) | |||||||
| 3 | -90.845 | -7.98179 | 4 | O(4) - Glu(336) | 3.10 3.10 | -91.302 | -0.45678 |
| O(4) - Thr(242) | 3.25 | ||||||
| O(8) - Gln(241) | 3.04 | ||||||
| O(12) - Ala(354) | |||||||
| 4 | -126.461 | -9.95729 | 4 | O(19) - Gln(241) | 3.11 3.10 | 15131 | 15257.5 |
| O(19) - Lys(467) | 2.94 | ||||||
| O(19) - Tyr(476) | 2.6 | ||||||
| O(17) - Tyr(476) | |||||||
| 5 | -105.783 | 0 | 0 | NIL | -99.66 | 6.12368 | |
| 6 | -145.405 | -13.0909 | 6 | O(4) - Lys(467) | 3.10 3.16 | 14108.4 | 14253.8 |
| O(4) - Gln(241) | 2.59 | ||||||
| O(4) - Tyr(476) | 3.39 | ||||||
| N(6) - Thr(242) | 3.06 | ||||||
| O(20) - Tyr(479) | 3.1 | ||||||
| O(22) - Tyr(479) | |||||||
| 7 | -145.198 | -9.96783 | 4 | O(10) - Gln(241) | 3.11 | 17158.4 | 17303.6 |
| O(10) - Lys(467) | 3.1 | ||||||
| O(10) - Tyr(476) | 2.21 | ||||||
| O(21) - Tyr(479) | 3.1 | ||||||
| 8 | -96.2771 | -1.96288 | 1 | O(4) - Glu(122) | 3.21 | -96.234 | 0.043551 |
| 9 | -140.301 | -2.98413 | 2 | O(9) - Gln(241) | 2.59 | 23260.2 | 23400.5 |
| O(9) - Tyr(479) | 3.49 | ||||||
| 10 | -124.438 | -2.87834 | 2 | O(8) - Gln(241) O(8) - Tyr(476) | 2.6 | 18207.5 | 18331.9 |
| 3.48 | |||||||
| Benazepril (Lotensin) | -75.071 | -4.74372 | 3 | 3.46 | -79.81 | -4.739 | |
| O(6) - His(343) | 3.1 | ||||||
| O(6) - Asp(375) | 3.12 | ||||||
| S(12) - His(313) | |||||||
| Captopril | -140.85 | -11.3733 | 5 | O(23) - Lys(467) | 2.82 | -145.62 | -4.773 |
| O(23) - Gln(241) | 2.92 | ||||||
| O(24) - Gln(241) | 3.32 | ||||||
| O(24) - Tyr(467) | 3.06 | ||||||
| O(6) - Asp(409) | 3.1 | ||||||
| Enalapril | -147.801 | -11.3185 | 6 | O(26) - His(347) | 3.22 | -154.79 | -6.986 |
| O(26) - Glu(344) | 3 | ||||||
| O(14) - Tyr(479) | 3.53 | ||||||
| O(27) – Tyr(479) | 3.25 | ||||||
| N(15) - Glu(344) | 2.58 | ||||||
| N(28) - Glu(122) | 2.62 | ||||||
| Lisinopril | -155.988 | -9.98388 | 4 | O(2) - Asn(237) | 2.77 | -161.16 | -5.168 |
| O(2) - Glu(336) | 3.1 | ||||||
| O(2) - Thr(242) | 3.1 | ||||||
| O(3) - Gln(241) | 2.7 |
Table 4: Docking results of predicted compounds (Table 2) with reference to the active drugs available in the market. Energy values are in kCal/mol.
ADME Results
Water solubility
It has been estimated that 40% of active new chemical entities (NCEs) used by different pharmaceutical companies are poorly water-soluble, i.e., these compounds exhibit an aqueous solubility <10 μM (5 μg/mL for a compound with a molecular weight of 500). When these poorly soluble NCEs are advanced further in discovery and are finally brought into development, they often turn out to be problematic due to incomplete absorption and low, inconsistent bioavailability [19]. Hence, an attempt has been made to design the proposed leads in a manner that they possess fair water solubility. They showed solubility comparable to the commercialized ones.
In order to predict Water Solubility, two different topological methods are included in Swiss ADME. The first one implements the ESOL model [20], and the second one is taken from Ali et al. [21]. Both are different on the basis of the seminal general solubility equation [22] since they avoid the melting point parameter; the latter being challenging to predict. Third predictor for solubility was developed by SILICOS-IT. All the predicted values are the decimal logarithm of the molar solubility in water (log S) [23]. A qualitative estimation of the class of solubility has been given according to the following log S scale: insoluble <−10 <poorly <−6 <moderately <−4 <soluble <−2 <very <0 <highly [23] as depicted in Table 5, the Proposed compounds are having good or comparable water solubility as compared with licensed drugs.
| S.NO | Log S (ESOL) | Solubility | Class | Log S (Ali) | Solubility | Class | Log S (SILICOS-IT) | Solubility | Class |
|---|---|---|---|---|---|---|---|---|---|
| 1 | -2.1 | 2.41e+00 mg/ml ; 7.90e-03 mol/l | Soluble | -1.62 | 7.27e+00 mg/ml ; 2.39e-02 mol/l | Very soluble | -0.57 | 8.12e+01 mg/ml ; 2.67e-01 mol/l | Soluble |
| 2 | -2.7 | 6.06e-01 mg/ml ; 2.02e-03 mol/l | Soluble | -2.24 | 1.73e+00 mg/ml ; 5.75e-03 mol/l | Soluble | -2.62 | 7.24e-01 mg/ml ; 2.41e-03 mol/l | Soluble |
| 3 | -2.1 | 2.57e+00 mg/ml ; 8.50e-03 mol/l | Soluble | -1.52 | 9.04e+00 mg/ml ; 2.99e-02 mol/l | Very soluble | -1.27 | 1.63e+01 mg/ml ; 5.38e-02 mol/l | Soluble |
| 4 | -1.2 | 1.56e+01 mg/ml ; 5.93e-02 mol/l | Very soluble | -0.64 | 6.02e+01 mg/ml ; 2.29e-01 mol/l | Very soluble | -0.84 | 3.82e+01 mg/ml ; 1.45e-01 mol/l | Soluble |
| 5 | -2.3 | 1.48e+00 mg/ml ; 4.90e-03 mol/l | Soluble | -1.92 | 3.65e+00 mg/ml ; 1.21e-02 mol/l | Very soluble | -1.27 | 1.63e+01 mg/ml ; 5.38e-02 mol/l | Soluble |
| 6 | -1.5 | 9.33e+00 mg/ml ; 3.06e-02 mol/l | Very soluble | -0.98 | 3.20e+01 mg/ml ; 1.05e-01 mol/l | Very soluble | -0.61 | 7.52e+01 mg/ml ; 2.47e-01 mol/l | Soluble |
| 7 | -2.1 | 2.67e+00 mg/ml ; 8.27e-03 mol/l | Soluble | -1.76 | 5.61e+00 mg/ml ; 1.74e-02 mol/l | Very soluble | -2.68 | 6.82e-01 mg/ml ; 2.11e-03 mol/l | Soluble |
| 8 | -3.7 | 6.23e-02 mg/ml ; 1.94e-04 mol/l | Soluble | -4.04 | 2.93e-02 mg/ml ; 9.15e-05 mol/l | Moderately soluble | -2.3 | 1.59e+00 mg/ml ; 4.97e-03 mol/l | Soluble |
| 9 | -2.6 | 8.67e-01 mg/ml ; 2.62e-03 mol/l | Soluble | -2.35 | 1.48e+00 mg/ml ; 4.47e-03 mol/l | Soluble | -3.13 | 2.42e-01 mg/ml ; 7.33e-04 mol/l | Soluble |
| 10 | -1.9 | 3.32e+00 mg/ml ; 1.15e-02 mol/l | Very soluble | -1.56 | 7.98e+00 mg/ml ; 2.76e-02 mol/l | Very soluble | -2.1 | 2.31e+00 mg/ml ; 7.98e-03 mol/l | Soluble |
| Benazepril | -2.9 | 5.44e-01 mg/ml ; 1.28e-03 mol/l | Soluble | -2.87 | 5.68e-01 mg/ml ; 1.34e-03 mol/l | Soluble | -6.09 | 3.47e-04 mg/ml ; 8.18e-07 mol/l | Poorly soluble |
| Captopril | -1.1 | 1.58e+01 mg/ml ; 7.29e-02 mol/l | Very soluble | -1.93 | 2.56e+00 mg/ml ; 1.18e-02 mol/l | Very soluble | -0.38 | 9.14e+01 mg/ml ; 4.21e-01 mol/l | Soluble |
| Elanopril | -1.6 | 1.02e+01 mg/ml ; 2.70e-02 mol/l | Very soluble | -1.49 | 1.21e+01 mg/ml ; 3.21e-02 mol/l | Very soluble | -3.7 | 7.46e-02 mg/ml ; 1.98e-04 mol/l | Soluble |
| Lisinopril | 0.15 | 5.76e+02 mg/ml ; 1.42e+00 mol/l | Highly soluble | 0.62 | 1.71e+03 mg/ml ; 4.21e+00 mol/l | Highly soluble | -3.44 | 1.46e-01 mg/ml ; 3.60e-04 mol/l | Soluble |
Table 5: Water solubility of the proposed compounds (Table 2) in comparison with the commercial ones.
Pharmacokinetics
Models compiled in the Pharmacokinetics section of Swiss ADME, finds out ADME behaviours of the molecules to be studied [23]. The first model is a multiple linear regression, that predicts the skin permeability coefficient (Kp)[24]. The predictions for passive human gastrointestinal absorption (HIA) and blood-brain barrier (BBB) permeation both can be studied from the BOILED-Egg model [25,26]. If we talk about the drug discovery processes, gastrointestinal absorption and brain access come across as important pharmacokinetic behaviours . The Brain Or Intestinal Estimate D permeation method (BOILED-Egg) is an accurately proposed model that serves the purpose of studying the possibility of a drug crossing the blood brain barrier by predicting its Lipophilicity and polarity [13]. Swiss ADME allows us to estimate if a chemical is a substrate of P-gp or inhibitor of certain CYP isoenzymes [23] which are CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP3A4 [24]. On inhibition, these isoenzymes can cause pharmacokinetics-related drug-drug interactions [27-29] which might lead adverse effects caused by poor clearance/accumulation of the drug or its metabolites [30]. P-gp (permeability glycoprotein) is key to estimate active efflux through biological membranes, like, from the gastrointestinal wall to the lumen or from the brain [25]. Another major role of P-gp is the protection of the central nervous system (CNS) from xenobiotics [26]. Hence it is important to estimate that a chemical is a substrate of P-gp or inhibitor of the most important CYP isoenzymes or not, as done in this case. Compounds 4, 6, 9, 10 (Table 6) are not showing such type of activities.
| S.no | GI absorption | BBB permeant | P-gp substrate | CYP1A2 inhibitor | CYP2C19 inhibitor | CYP2C9 inhibitor | CYP2D6 inhibitor | CYP3A4 inhibitor | Log Kp (skin permeation) (in cm/s) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | High | No | Yes | No | No | No | No | No | 7.66 |
| 2 | High | No | Yes | No | No | No | Yes | No | 7.17 |
| 3 | High | No | Yes | No | No | No | No | No | 7.67 |
| 4 | High | No | No | No | No | No | No | No | 8.03 |
| 5 | High | No | Yes | No | No | No | No | No | 7.40 |
| 6 | High | No | No | No | No | No | No | No | 8.10 |
| 7 | High | No | Yes | No | No | No | No | No | 7.92 |
| 8 | High | No | Yes | No | No | No | Yes | No | 6.30 |
| 9 | High | No | No | No | No | No | No | No | 7.52 |
| 10 | High | No | No | No | No | No | No | No | 7.81 |
| Benazopril | High | No | Yes | No | No | No | Yes | Yes | 7.99 |
| Captopril | High | No | No | No | No | No | No | No | 7.38 |
| Elanopril | High | No | Yes | No | No | No | No | No | 8.65 |
| Lisinopril | High | No | Yes | No | No | No | No | No | 10.80 |
Table 6: Pharmacokinetics of the proposed compounds (Table 2) in comparison with the commercial ones
Drug-likeliness
"Drug-likeness" qualitatively evaluates the chance of a molecule to become an oral drug with respect to bioavailability [23]. This section gives access to five different rule-based filters [23]. The Lipinski rule-of-five is as described in ref [31] along with MLOGP acting as lipophilicity threshold whose value must be lesser than 4.15. The Ghose filter has been detailed in the original publication [32]. The Veber filter is implemented from the seminal paper [33]. The Egan filter has been adopted from from the Egan Egg [34-37], and the closed-source ALOGP98 was replaced by WLOGP [25]. The Muegge filter [35] was employed to fit Swiss ADME implementation and usage by the calculation of XLOGP3 as lipophilicity descriptor. The Bioavailability Score was adapted from Martin et al. [36]. No violation of any rule described here appears in the case of proposed compounds contrary to the commercialized drugs that seem to show certain violations as depicted in Table 7
| S.no | Lipinski | Ghose | Veber | Egan | Muegge | Bioavailability Score |
|---|---|---|---|---|---|---|
| 1 | Yes | Yes | Yes | Yes | Yes | 0.55 |
| 2 | Yes | Yes | Yes | Yes | Yes | 0.55 |
| 3 | Yes | Yes | Yes | Yes | Yes | 0.55 |
| 4 | Yes | Yes | Yes | Yes | Yes | 0.55 |
| 5 | Yes | Yes | Yes | Yes | Yes | 0.55 |
| 6 | Yes | Yes | Yes | Yes | Yes | 0.55 |
| 7 | Yes | Yes | Yes | Yes | Yes | 0.55 |
| 8 | Yes | Yes | Yes | Yes | Yes | 0.55 |
| 9 | Yes | Yes | Yes | Yes | Yes | 0.55 |
| 10 | Yes | Yes | Yes | Yes | Yes | 0.55 |
| Benazopril | Yes | Yes | Yes | Yes | \Yes | 0.55 |
| Captopril | Yes | Yes | Yes | Yes | Yes | 0.56 |
| Elanopril | Yes | Yes | No; 1 violation: Rotors>10 | Yes | Yes | 0.55 |
| Lisinopril | Yes | Yes | No; 1 violation: Rotors>10 | No; 1 violation: TPSA>131.6 | No; 1 violation: XLOGP3<-2 | 0.55 |
Table 7: Drug likeness of the proposed compounds (Table 2) in comparison with the commercial ones.
Medicinal Chemistry
Lead optimization is an important filter from which the proposed molecules (Table 2) has to passed through [23,38]. Two methods are employed in this section - PAINS (for pan assay interference compounds) and Brenk filters [38,39]. Synthetic Accessibility (SA) Score suggests us the ease of synthesis [23]. The score could be between 1 to 10 where 1 is very easy while 10 denotes difficulty in synthesizing. Hence, Table 8 has been shown, representing all of the above-mentioned filters. The proposed drugs show better results in terms of lead likeliness as compared to commercial drugs. Results suggests that it will possible to synthesized the proposed compounds with as much difficulty as the commercial drugs.
| S.no | PAINS | Brenk | Leadlikeness | Synthetic accessibility |
|---|---|---|---|---|
| 1 | 0 alert | 0 alert | Yes | 4.93 |
| 2 | 0 alert | 0 alert | Yes | 3.34 |
| 3 | 0 alert | 0 alert | Yes | 4.52 |
| 4 | 0 alert | 0 alert | Yes | 4.63 |
| 5 | 0 alert | 0 alert | Yes | 4.54 |
| 6 | 0 alert | 0 alert | Yes | 4.9 |
| 7 | 0 alert | 0 alert | Yes | 3.82 |
| 8 | 0 alert | 0 alert | Yes | 4.43 |
| 9 | 0 alert | 0 alert | Yes | 3.74 |
| 10 | 0 alert | 0 alert | Yes | 3.5 |
| Benazopril | 0 alert | 0 alert | No; 2 violations: MW>350, Rotors>7 | 4.00 |
| Captopril | 0 alert | 1 alert: thiol_2 | No; 1 violation: MW<250 | 2.47 |
| Elanopril | 0 alert | 0 alert | No; 2 violations: MW>350, Rotors>7 | 3.61 |
| Lisinopril | 0 alert | 0 alert | No; 2 violations: MW>350, Rotors>7 | 3.67 |
Table 8: Medicinal Chemistry of the proposed compounds (Table 2) in comparison with the commercial ones.
Lipophilicity
The partition coefficient (log Po/w) is the measure of Lipophilicity of a drug. Swiss ADME provides access to five easily available predictive models. The first one is XLOGP3 which is an atomistic method adopted for the calculation of log P [40]; Second is WLOGP which is the implementation of an atomistic method that in turn is based upon the fragmental system suggested by Wildman and Crippen [41]; The third one is MLOGP which is a prototype of topological method that relies on a linear relationship with 13 different molecular descriptors. [42-44] and the fourth one is SILICOS-IT which is a hybrid method that is based upon 27 fragments and 7 topological descriptors; and lastly, ilogP [23]. Swiss ADME finally provides a consensus log Po/w value, which is the arithmetic mean of the above-mentioned predictive values [23]. The results (Table 9) are comparable to commercialized drugs.
| S.no | Log Po/w (iLOGP) | Log Po/w (XLOGP3) | Log Po/w (WLOGP) | Log Po/w (MLOGP) | Log Po/w (SILICOS-IT) | Consensus Log Po/w |
|---|---|---|---|---|---|---|
| 1 | 2.68 | 0.7 | 0.37 | 1.28 | 0.18 | 1.04 |
| 2 | 2.56 | 1.36 | 0.79 | 1.64 | 1.11 | 1.49 |
| 3 | 2.23 | 0.67 | 0.23 | 0.78 | 0.77 | 0.94 |
| 4 | 2 | -0.18 | 0.27 | 0.8 | 0.85 | 0.75 |
| 5 | 2.67 | 1.05 | 0.58 | 1.19 | 0.77 | 1.25 |
| 6 | 2.21 | 0.08 | 0.04 | 0.38 | 0.69 | 0.68 |
| 7 | 1.97 | 0.49 | -0.08 | 0.5 | 1.88 | 0.95 |
| 8 | 2.7 | 2.76 | 1.81 | 2.08 | 1.45 | 2.16 |
| 9 | 2.38 | 1.12 | 0.38 | 0.98 | 1.45 | 1.26 |
| 10 | 1.78 | 0.36 | 0 | 0.72 | 1.08 | 0.79 |
| Benazepril | 3.05 | 1.26 | 2.19 | 2.23 | 3.14 | 2.37 |
| Captopril | 1.44 | 0.34 | 0.25 | 0.45 | 0.61 | 0.62 |
| Elanopril | 3.08 | -0.07 | 1.22 | 1.32 | 2.22 | 1.55 |
| Lisinopril | 2.44 | -2.86 | 0.85 | -1.46 | 1.65 | 0.13 |
Table 9: Lipophilicity of the proposed compounds (Table 2) in comparison with the commercial ones.
Toxicity
Table 10 shows the toxicity prediction of all the proposed compounds. Hepatotoxicity, cytotoxicity, carcinogenicity, mutagenicity and immunotoxicity of each of these compounds have been found to be completely or almost inactive. Our livers are important targets of the toxicity of drugs, xenobiotics, and oxidative stress. Hence, it is important for the leads to have minimum hepatotoxicity, as shown by the proposed compounds [14]. Carcinogenicity is another toxicological endpoint causing the highest concern [15] making it an important factor to be taken care of, hence the compounds designed show no carcinogenicity. Immune-related drug responses are very common sources of idiosyncratic toxicity [16]; hence an effort has been made to design the compounds that show minimum immunotoxicity.
| S.no | Hepatotoxicity Prediction | Carcinogenicity Prediction | Immunotoxicity Prediction | Mutagenicity Prediction | Cytotoxicity Prediction |
|---|---|---|---|---|---|
| 1 | Inactive | Inactive | Inactive | Inactive | Inactive |
| 2 | Inactive | Inactive | Inactive | Inactive | Inactive |
| 3 | Inactive | Inactive | Inactive | Inactive | Inactive |
| 4 | Inactive | Slightly active | Inactive | Inactive | Inactive |
| 5 | Inactive | Inactive | Inactive | Inactive | Inactive |
| 6 | Inactive | Inactive | Inactive | Inactive | Inactive |
| 7 | Inactive | Inactive | Inactive | Inactive | Slightly active |
| 8 | Inactive | Inactive | Inactive | Inactive | Inactive |
| 9 | Inactive | Inactive | Inactive | Inactive | Inactive |
| 10 | Inactive | Inactive | Slightly active | Inactive | Slightly active |
| Benazopril | Inactive | Inactive | Inactive | Inactive | Inactive |
| Captopril | Inactive | Active | Inactive | Inactive | Inactive |
| Elanopril | Inactive | Inactive | Inactive | Inactive | Inactive |
| Lisinopril | Inactive | Inactive | Inactive | Inactive | Inactive |
Table 10: Toxicity of the predicted compounds in comparison with the commercial ones.
Conclusion
The study shows that new compounds proposed shown in Table 2 predicted higher activity than the reported compounds shown in Table 1. The docking results of the proposed compounds are comparable to the commercial drugs, whereas the ADME properties and the toxicity prediction of the proposed compounds are better than licensed compounds. The Synthetic accessibility of the proposed compounds is also comparable to the licensed compounds which shows that they can be synthesized synthetically just like licensed compounds; these compounds can hence be synthesized and tested in the wet lab.
Acknowledgements
The authors are thankful to the Meerut Institute of Engineering and Technology for providing the infrastructure and support for completion of this project. This work was also supported by the Molexus. Our profound gratitude is expressed to Mr. René Thomsen for providing the academic License of MVD software used for Docking. We acknowledge Chem Axon Ltd. (www.chemaxon.com) for the academic license agreement. ADMET properties were studied by the help of Swiss ADME and Pro Tox-ll.
Conflicts of Interests
The author has no conflict of interests.
References
- D Coates. Int J Biochem Cell Biol. 2003, 35(6): p. 769-73.
- LJ Dell'Italia, P Rocic and PA Lucchesi. Cur Prob in Cardiology. 2002, 27(1): p. 6-36.
- NJ Brown and DE Vaughan. 1998, 97(14): p. 1411-1420.
- T Münzel and JF Keaney. 2001, 104(13): p. 1571-1574.
- NJ Brown and DE Vaughan. 1998, 97(14): p. 1411-1420.
- C Isarankura-Na-Ayudhya, T Naenna, C Nantasenamat et al., EXCLI J. 2009, 8: p. 74-88.
- T Wang, MB Wu, JP Lin and LR Yang, Exp Opion Drug Dis. 2015, 10(12): p. 1283-1300.
- D Addla, A Jallapally, A.Kanwal et al., Bio Med chem. 2013, 21(15): p. 4485-4493.
- MN Drwal, P Banerjee, M Dunkel et al., Nuclacid Res. 2014, 42(W1): p. 53-58.
- PW Kenny and CA Montanari. J Comp-aid Mol Des. 2013, 27(1): p. 1-13.
- A Daina and V Zoete. Chem Med Chem. 2016, 11(11): p. 1117-1121.
- H Jaeschke, GJ Gores, AI Cederbaum et al., Toxico Sci. 2002, 65(2): p. 166-176.
- S Kar and K Roy. Ind J Biochem Biophy. 2011, 48(2): p. 111-22.
- J Descotes. Immunotoxicity of drugs and chemicals. 1986.
- N Agarwal, A Bajpai, V Srivastava et al., Structural Biology, 2013, 810691: p. 11.
- ChemDes,http://www.scbdd.com/chemdes/
- Toxicity prediction server. http://tox.charite.de/protox_II/index.php?site=compound_input
- ADME and druglikeness properties evaluation server.http://www.swissadme.ch/
- T Heimbach, D Fleisher, and A Kaddoumi. Prodrugs. 2007, p. 157-215.
- JSD ESOL. J Chem Inf Model. 2004, 44: p. 1000-1005 .
- J Ali,P Camilleri ,MB Brown et al., J. Chem. Inf. Model,2012, 52: p. 420–428.
- ]SH Yalkowsky &Valvani SC. J Pharm Sci. 1980, 69: p. 912-922.
- A Daina, O Michielin and V Zoete. Scientific reports, 2017, 7: p. 42717.
- RO Potts and RH Guy. Pharm Res 1992, 09: p. 663-669.
- A Daina and V Zoete. ChemMedChem, 2016, 11:p. 1117–1121.
- F Montanari and GF Ecker. Adv Drug Deliv Rev. 2015, 86: p. 17-26.
- G Szakács, A Váradi, C Ozvegy-Laczka et al., Drug Discov Today. 2008, 13: p. 379-393.
- PF Hollenberg. Drug Metab Rev. 2002, 34: p. 17-35.
- SM Huang et al. J Clin Pharmacol. 2008, 48: p. 662-670.
- J Kirchmair et al., Nature Rev Drug Discov. 2015,14: p. 387-404.
- CA Lipinski, F Lombardo, BW Dominy et al., Adv Drug Deliv Rev. 2001, 46: p. 3-26.
- AK Ghose, VNViswanadhan and JJ Wendoloski. J Comb Chem 1999, 1: p. 55-68.
- DF Veberet al., J Med Chem. 2002, 45: p. 2615-2623.
- WJ Egan, KM Merz and JJ Baldwin. J Med Chem. 2000, 43: p. 3867-3877.
- I Muegge, SL Heald and D Brittelli. J Med Chem. 2001, 44: p. 1841-1846.
- YC Martin. J Med Chem. 2005, 48:p. 3164-3170.
- WJ Egan and G Lauri. Adv Drug Deliv. 2002, Rev.54: p. 273-289.
- S Teague, A Davis, P Leeson et al., Angew Chem Int Ed Engl. 1999, 38: p. 3743-3748.
- R Brenk et al., Chem Med Chem. 2008, 3: p. 435-444.
- JB Baell and GA Holloway. J Med Chem. 2010, 53: p. 2719-2740.
- ]T Cheng et al., J Chem Inf Model. 2007, 47: p. 2140-2148.
- SA Wildman and GM Crippen. J Chem Inf Model. 1999, 39: p. 868-873.
- I Moriguchi, H Shuichi, Q Liu et al., Chem Pharm Bull. 1992, 40: p. 127-130.
- I Moriguchi, H Shuichi, I Nakagome et al., Chem Pharm Bull. 1994, 42: p. 976-978.
- S Kusumaningrum, E Budianto, S Kosela et al., J App Sci. 2014, 4: p. 47-53.



