Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 6
Process for the Preparation of Dolutegravir Intermediate: (R)-3-Amino Butanol
Sureshbabu Jayachandra, Madhuresh K Sethi*, Vipinkumar Kaushik, Vijayakrishna Ravi, Sanjay Mahajan, Bhairaiah Mara, Gurleen Kaur and Purbita Chakraborty
R & D, Mylan Laboratories Ltd., Plot No. 31, 32, 33 and 34 A ANRICH Industrial Estate, Bollaram (Village), Jinnaram (Mandal), Medak (District) 502325, Telangana, India
- *Corresponding Author:
- Madhuresh K Sethi
R & D, Mylan Laboratories Ltd.
Plot No. 31, 32, 33 and 34 A ANRICH Industrial Estate
Bollaram (Village), Jinnaram (Mandal), Medak (District) 502325, Telangana, India
Abstract
Dolutegravir, a second-generation HIV Integrase Strand Transfer Inhibitor (INSTI), is the most current antiretroviral ratified for treatment of HIV-1 infection. This approval is based on the superiority of Dolutegravir in clinical trials over the well-known antiretroviral marketed drugs like Efavirenz, Raltegravir, and Darunavir. Largely, Dolutegravir has established excellent tolerability, limited drug interactions, insignificant drug resistance and once-daily dosing for treatment-naive patients. (R)-3-amino butanol is a key unit for the synthesis of Dolutegravir and rarer methods of synthesis are mentioned in the literature.
Keywords
Integrase strand transfer inhibitor, Antiretroviral drugs
Introduction
For nearly two epochs, morbidity and mortality of HIV infected patients has declined because of antiretroviral therapy. Antiretrovirals work by inhibiting HIV integrase by binding to the integrase active site and blocking the strand transfer step of retroviral Deoxyribonucleic Acid (DNA) integration which is vital for the HIV replication cycle [1]. Out of five known distinct classes of antiretroviral medications, HIV integrase strand transfer inhibitors (INSTIs) have had a huge impact on the treatment of HIV infection. There are currently three US FDA approved INSTIs: Raltegravir (Isentress®), Elvitegravir (Stribild®) and Dolutegravir (Tivicay®) [2].
Dolutegravir (DTG) (Figure 1), a second-generation INSTI, an FDA-approved drug on August 13, 2013 for the treatment of HIV infection, is an integrase inhibitor marketed as Tivicay by GlaxoSmithKline (GSK) [2]. On January 16, 2014, it was approved by the European Commission for use all over the European Union. It has proven remarkable antiviral value based on randomized controlled trials when compared to other first-line regimens, acclaimed by the Department of Health and Human Services HIV treatment guidelines for adults and adolescents.
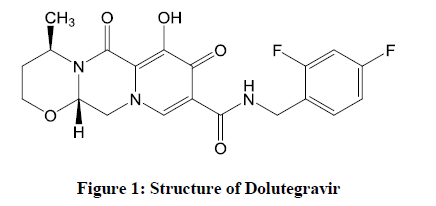
Figure 1: Structure of Dolutegravir
Although (R)-3-amino butanol is a key unit for the synthesis of Dolutegravir (Figure 2), fewer routes of synthesis are described in the literature [3]. This paper will shortlist all the possible routes mentioned in the literature and will provide an improved procedure suggested or undergone in our lab.
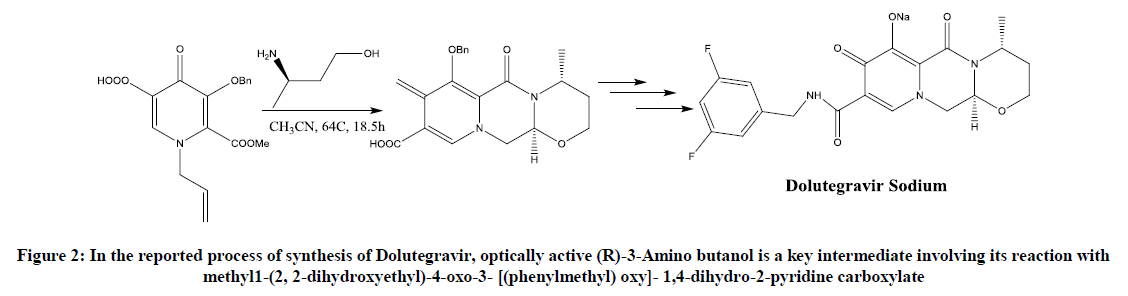
Figure 2: In the reported process of synthesis of Dolutegravir, optically active (R)-3-Amino butanol is a key intermediate involving its reaction with methyl1-(2, 2-dihydroxyethyl)-4-oxo-3- [(phenylmethyl) oxy]- 1,4-dihydro-2-pyridine carboxylate
Materials and Methods
Catalytic methods
Describe the various methods in the literature that been reported towards the synthesis of (R)-3-amino butanol using chemical catalysts. It is to be noted that all the listed methodologies for the synthesis of this intermediate employs tedious chiral synthetic precursors, expensive ruthenium/rhodium catalysts, punitive reaction conditions, harmful reagents, raised temperatures and lengthy reaction times which are incongruous for enormous scale set-ups.
Method-1
This method is reported by Jing vii for the synthesis of (R)-3-aminobutanol (Figure 3) [4].
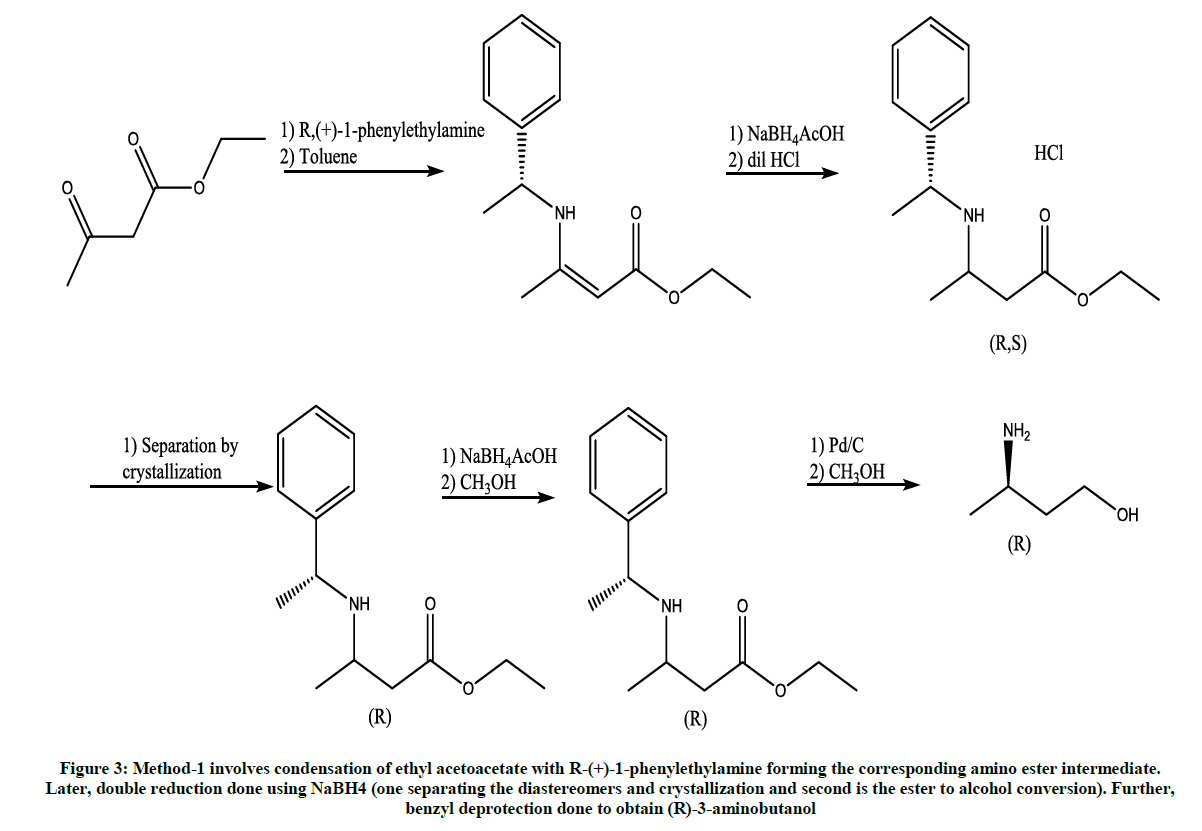
Figure 3: Method-1 involves condensation of ethyl acetoacetate with R-(+)-1-phenylethylamine forming the corresponding amino ester intermediate. Later, double reduction done using NaBH4 (one separating the diastereomers and crystallization and second is the ester to alcohol conversion). Further, benzyl deprotection done to obtain (R)-3-aminobutanol
Main disadvantage of this method is the very low yields and usage of expensive ‘Palladium’ catalyst.
Method-2
This method involves asymmetric synthesis reaction (Figure 4) [5].
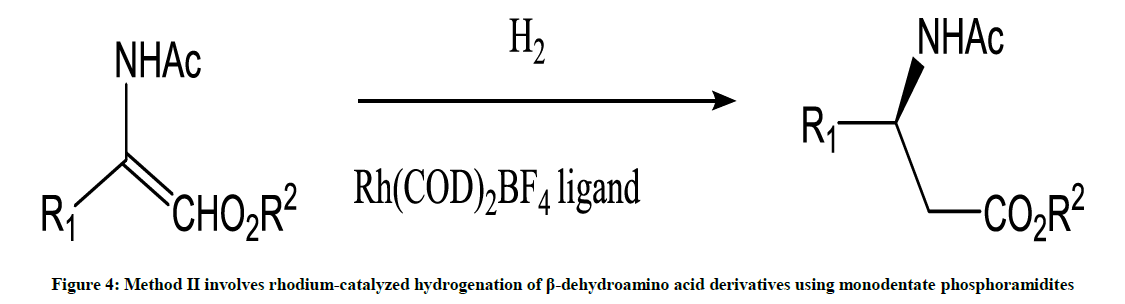
Figure 4: Method II involves rhodium-catalyzed hydrogenation of β-dehydroamino acid derivatives using monodentate phosphoramidites
Method-3
This is a new catalytic route to β-3-amino acids (Figure 5) [6].
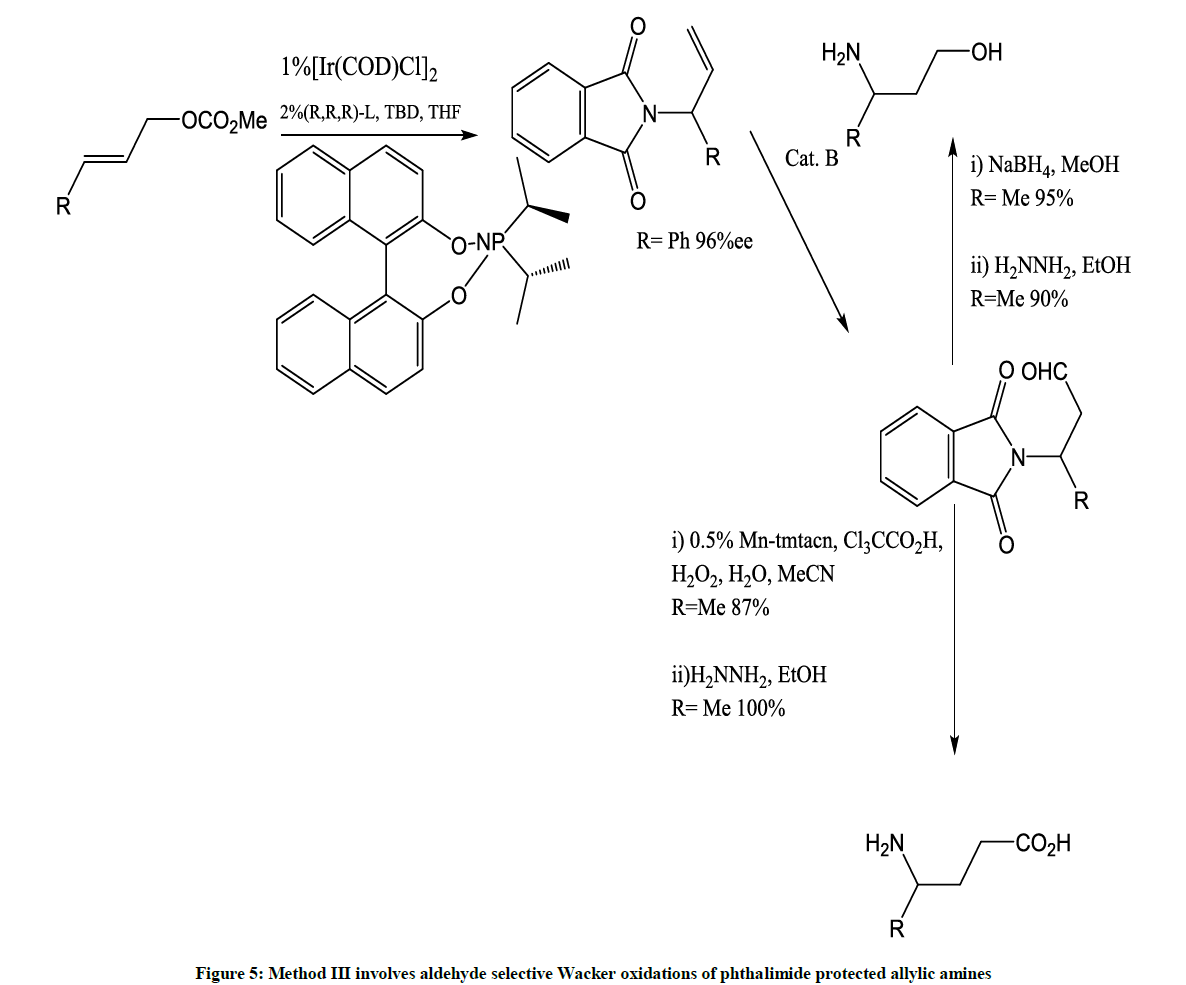
Figure 5: Method III involves aldehyde selective Wacker oxidations of phthalimide protected allylic amines
Method-4
This method involves an enantioselective conjugate addition (Figure 6) [7].
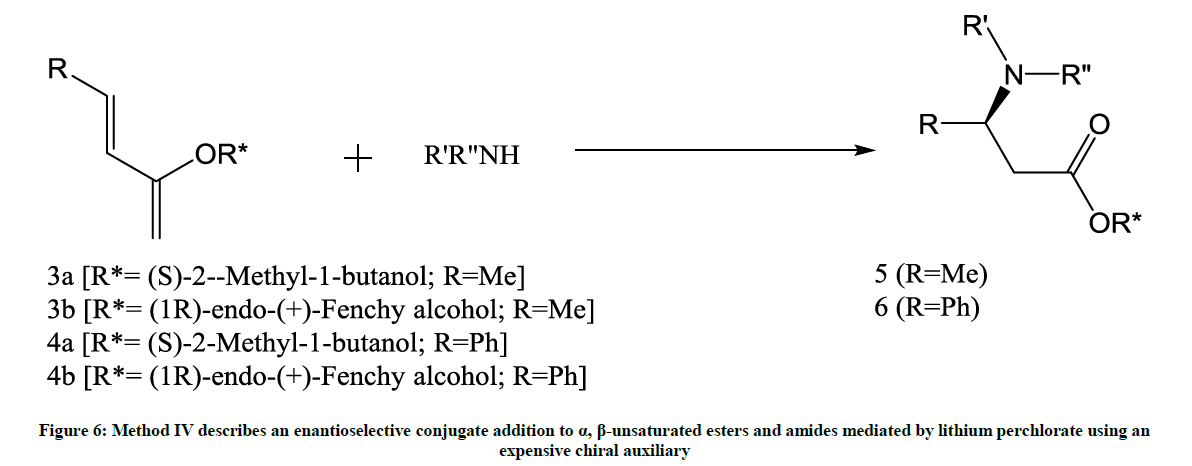
Figure 6: Method IV describes an enantioselective conjugate addition to α, β-unsaturated esters and amides mediated by lithium perchlorate using an expensive chiral auxiliary
Method-5
This method is for the synthesis of N-unprotected β-amino esters (Figure 7) [8].
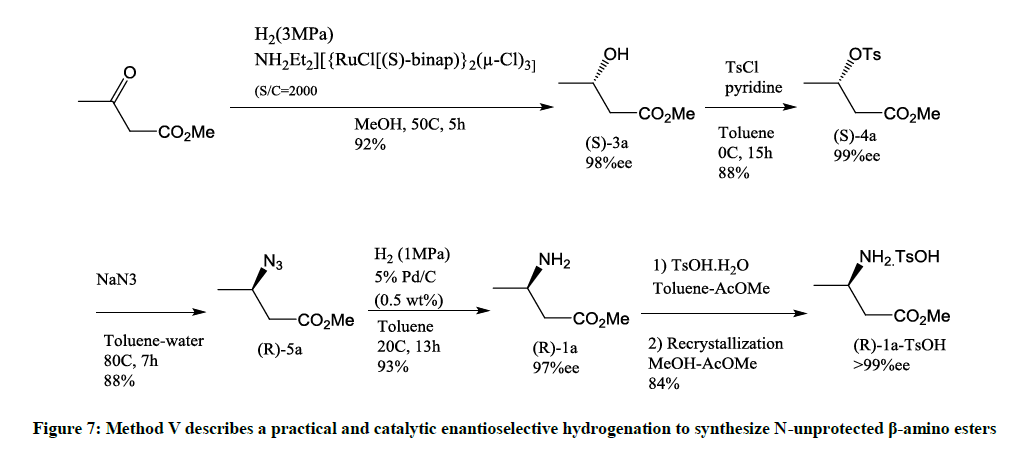
Figure 7: Method V describes a practical and catalytic enantioselective hydrogenation to synthesize N-unprotected β-amino esters
Enzymatic methods
Describe selective synthesis of (R)-3-amino butanol with the help of biodegradable and eco-friendly biological enzymes. These methods ensure minimal use of chemicals and no other byproduct formation during the reaction (only R and S).
Method 1
Involving the use of the enzyme Penicillin Acylase G (Figure 8)
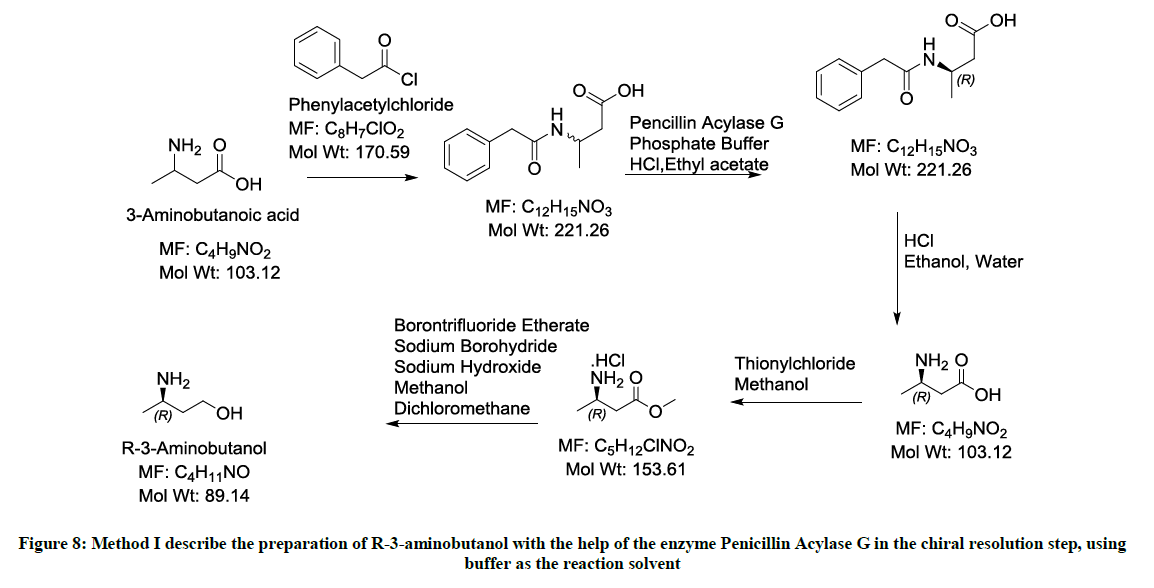
Figure 8: Method I describe the preparation of R-3-aminobutanol with the help of the enzyme Penicillin Acylase G in the chiral resolution step, using buffer as the reaction solvent
Method 2
Involving the use of a recombinant enzyme (Figure 9)
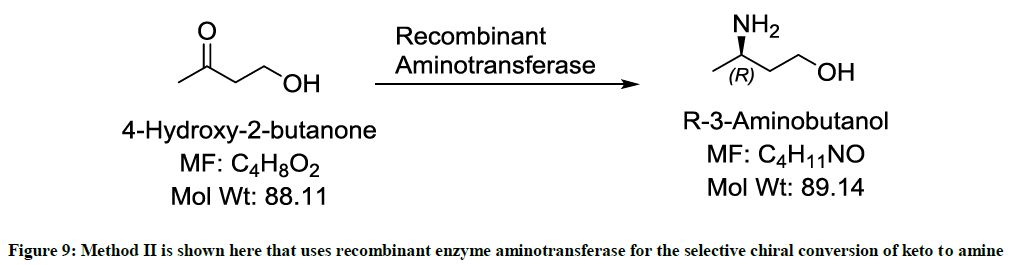
Figure 9: Method II is shown here that uses recombinant enzyme aminotransferase for the selective chiral conversion of keto to amine
Method 3
Involving the use of Yeast cells (Figure 10)
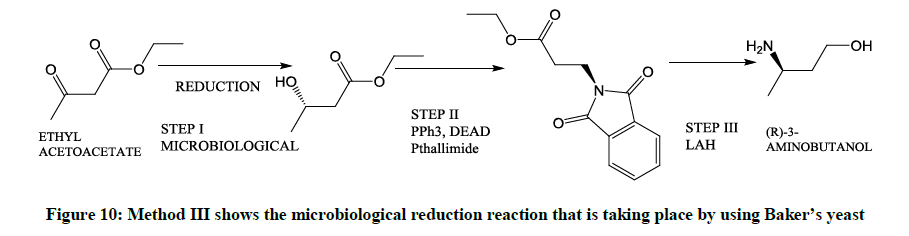
Figure 10: Method III shows the microbiological reduction reaction that is taking place by using Baker’s yeast
Method 4
Involving the enzyme ene-reductase (Figure 11)
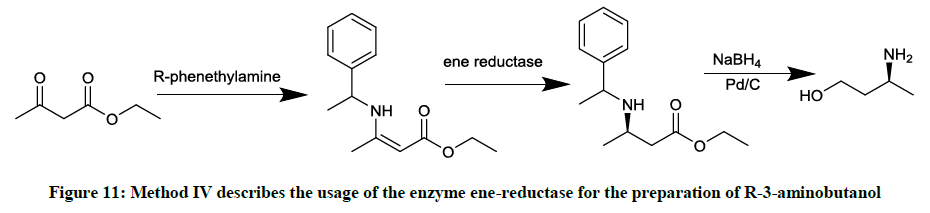
Figure 11: Method IV describes the usage of the enzyme ene-reductase for the preparation of R-3-aminobutanol
METHOD-V
Involving a lipase enzyme (Figure 12)
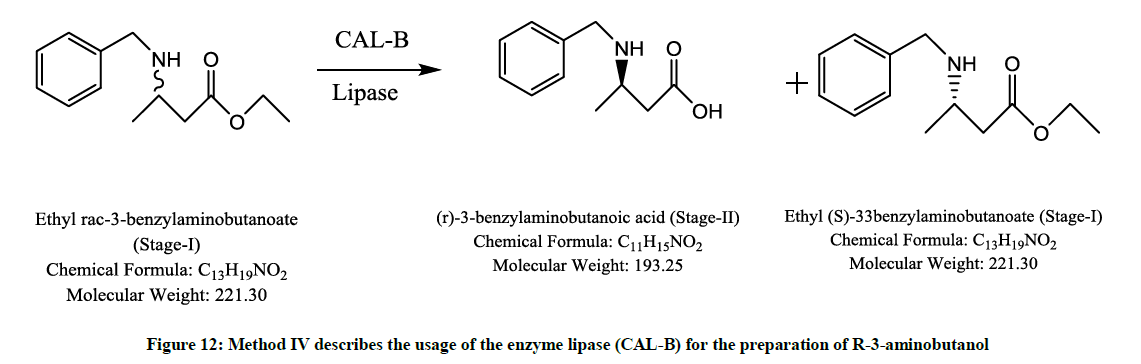
Figure 12: Method IV describes the usage of the enzyme lipase (CAL-B) for the preparation of R-3-aminobutanol
Resolution methods
Describe a known chemical method used for resolution of 3-aminobutanol to R-3-aminobutanol (Figure 13)
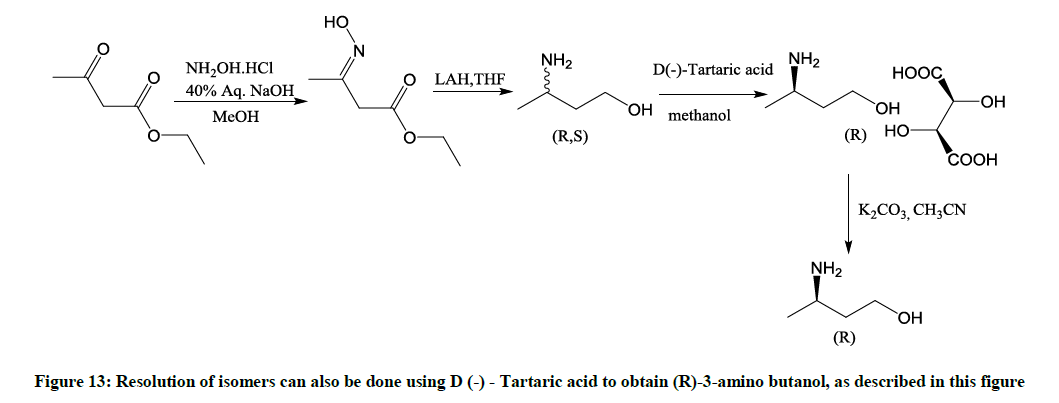
Figure 13: Resolution of isomers can also be done using D (-) - Tartaric acid to obtain (R)-3-amino butanol, as described in this figure
So, in the existent investigation, an endeavor is explored to develop an efficient, inexpensive and convenient route for the synthesis of (R)-3- aminobutanol.
Illustrates the findings and in-depth process of a five-steps preparation technique of (R)-3- amino butanol (Schemes 1-5) as performed by our team.
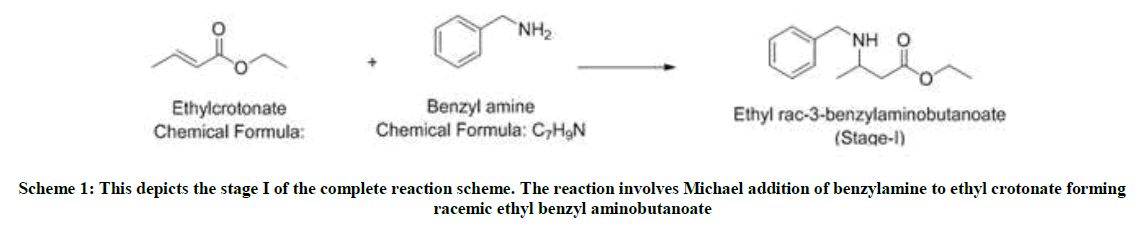
Scheme 1: This depicts the stage I of the complete reaction scheme. The reaction involves Michael addition of benzylamine to ethyl crotonate forming racemic ethyl benzyl aminobutanoate
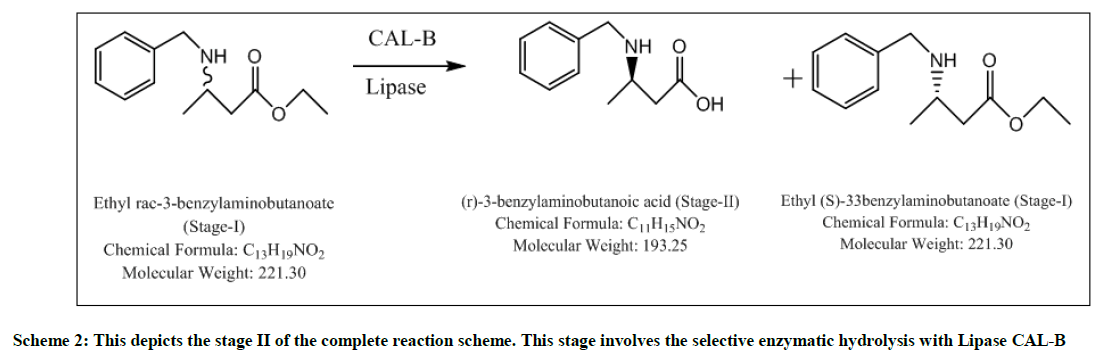
Scheme 2: This depicts the stage II of the complete reaction scheme. This stage involves the selective enzymatic hydrolysis with Lipase CAL-B
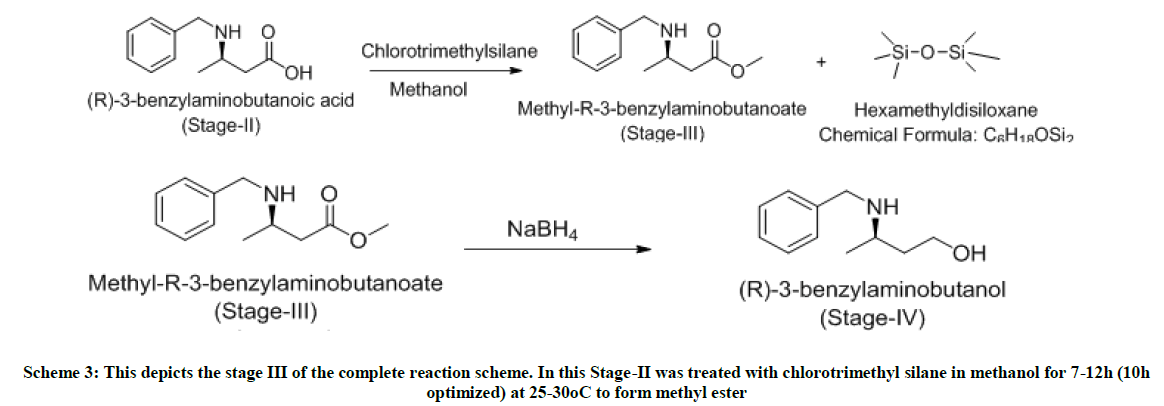
Scheme 3: This depicts the stage III of the complete reaction scheme. In this Stage-II was treated with chlorotrimethyl silane in methanol for 7-12h (10h optimized) at 25-30°C to form methyl ester
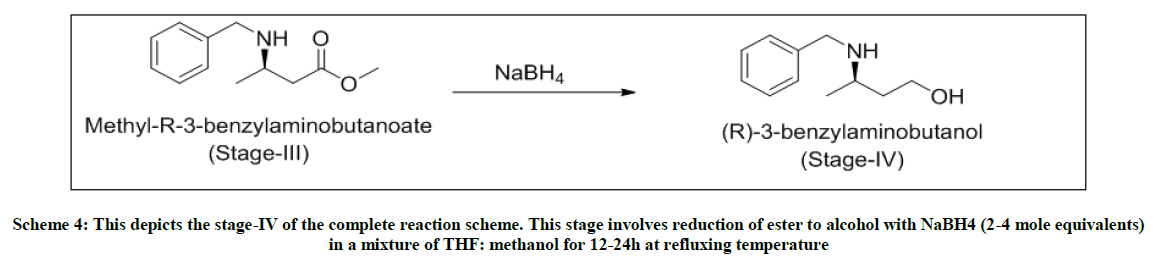
Scheme 4: This depicts the stage-IV of the complete reaction scheme. This stage involves reduction of ester to alcohol with NaBH4 (2-4 mole equivalents) in a mixture of THF: methanol for 12-24h at refluxing temperature
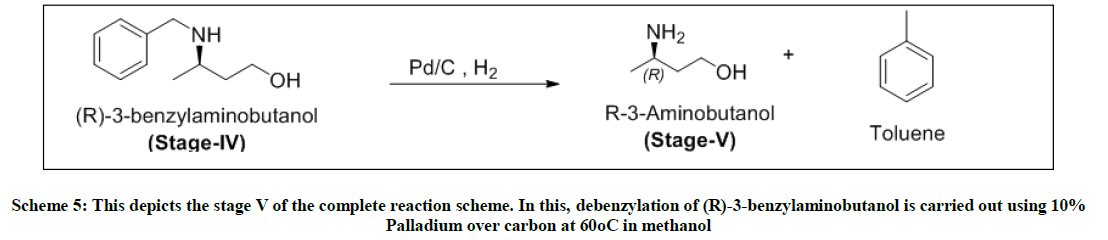
Scheme 5: This depicts the stage V of the complete reaction scheme. In this, debenzylation of (R)-3-benzylaminobutanol is carried out using 10% Palladium over carbon at 60°C in methanol
To develop improved and convenient selective route for the synthesis of (R)-3- amino butanol using economical, cheap and easily available starting materials, eco-friendly reagents, simple reaction procedures, minimal chemical steps, lesser reaction time, no harsh temperatures, easy workups, cost effective overall process giving higher yields, greater chiral and HPLC purity, the following five steps procedure performed in our lab will be discussed in detail in this manuscript.
For Stage-I, number of chemical experiments were conducted to optimize the favorable process that includes:
i. Variation of solvents (with (acetonitrile) or without solvent)
ii. Variation of temperatures (60-90°C)
iii. Variation in the reaction timings (24 h to 72 h)
iv. With and without Catalyst- DBU (Diaza bicyclo undecane)
v. With and without Enzyme-CAL-B
vi. Used in the next step with and without workup
vii. Used with or without further purification (column chromatography or fractional distillation)
viii. Adjusting the pH with 30% NaOH or NH3 solution (workup)
The possible impurities formed in the stage-1 (Figure 14) are as follows:
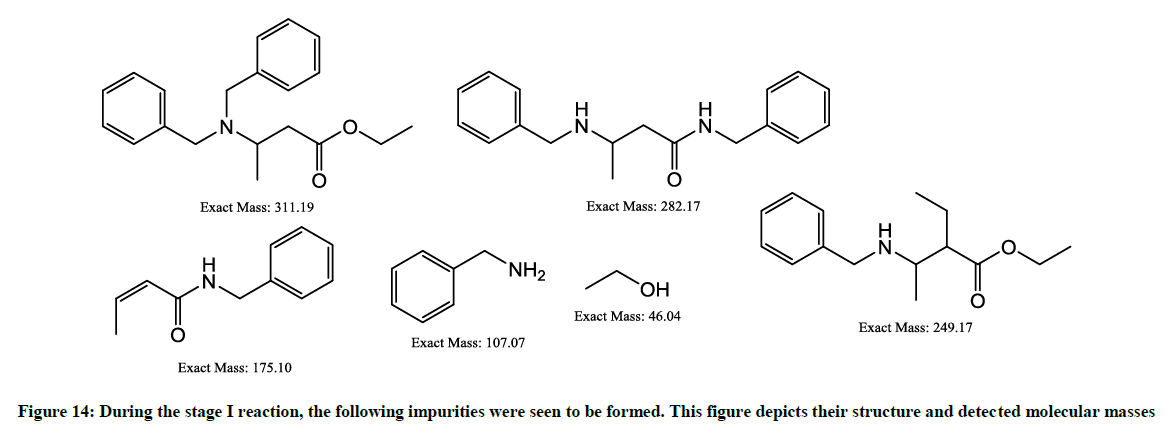
Figure 14: During the stage I reaction, the following impurities were seen to be formed. This figure depicts their structure and detected molecular masses
The major advantages of enzymatic step or Stage-II are its selectivity, eco-friendly nature, being easily decomposed, higher chiral purity, inexpensive when compared to chiral reagents, chiral catalysts and chiral auxiliaries. The reaction mixture was screened with different enzymes like CAL-B (Fermenta), TL Lipase liquid, TL Lipase Immobilized, CAL-BY-T2-150, Novozyme 51032, Lipase from CRL. Screening is done using CAL- B which is a hit and the reaction is optimized using various solvents like THF, Toluene, 1-butanol, t-pentanol and water. In Methanol, 1, 4- dioxane, no selectivity obtained but with recycled/ recovered enzymes, selective product formed. S-ester is formed as a byproduct which can be racemized and can be reutilized. Yields are quantitative, HPLC purity is above 90% and chiral purity which can still be improved is above 85%. In Stage-III, selectivity is retained, quantitative yield, easy layer separations, higher HPLC purity (99%), impurities (mostly water soluble) removed in water workup. At Stage-IV, impurities formed (mostly water soluble) can be removed in water workup. This reaction results in quantitative yield and higher purity. At the end of Stage-V chiral purity is 88% and HPLC purity is 87%, which was significantly improved to 98% post fractional distillation.
Experimental
General
Melting points were determined by open capillary using a Veego Programmable Melting/Boiling Point Apparatus and are uncorrected. IR spectra were recorded on Perkin Elmer FT-IR Spectrometer Spectrum Two. Molecular masses and HPLC purity were determined by Agilent LCMS constituting LC 1260 infinity and MS SQD 6120. 1H-NMR and 13C-NMR were recorded on Bruker 500 (125 MHz) spectrometer in deuterated chloroform and deuterated methanol. Chemical shift values are expressed in terms of parts per million and J values are in Hertz. Splitting patterns are abbreviated as: s- singlet, d- doublet, dd- double doublet, t- triplet and m- multiplet.
Stage-I-Michael addition
Ethyl crotonate (1.0 mole eq) and benzylamine (1.1 mole eq) are charged in a 4-neck flask and stirred for 12 h 80°C. The reaction is monitored using thin layer chromatography (TLC) and HPLC. After the reaction is complete, the reaction mixture is degassed under vacuum at 75°C. To the reaction mixture, added ethyl acetate (10 volumes) and water (10 volumes) and cooled it. At 0-5°C, the pH is slowly adjusted to zero using 1: 1 HCl solution. Settled and separated the layers and washed the aqueous layer twice with ethyl acetate (2 volumes). Then again charged the aqueous layer in a flask and fresh ethyl acetate (5 volumes) is charged. On cooling the mixture to 0-5°C, slowly adjusted the pH to 9 using liquid NH3. After settling and separating the layers, Organic layer washed twice with water (5 volumes) and dried over sodium sulfate. The ethyl acetate is distilled under vacuum to obtain yellow oil. Yield: 1.4 w/w, Mass: 222 (m+1), HPLC purity: 90%.
Stage-II-Selective enzymatic hydrolysis
To a solution of t-pentanol (2.5 volumes), added 10% enzyme CAL-B, and the starting material (stage-I) (1mole eq) and stirred slowly for 4 hours at 25-30°C. On the completion of the reaction, filtered the enzyme through celite and washed it with t-pentanol (2 volumes). Distilled off t-pentanol under vacuum, at 45°C. To the reaction mixture, charged MTBE (5 volumes) and water (5 volumes). Stirred, settled and separated the layers. Aqueous layer extracted twice with MTBE (2 volumes). Organic layer dried over sodium sulfate and then distilled off under vacuum at 40°C to yield selective R-isomer. SOR: -27.42. Yield: 0.4 w/w, Mass: 194 (m+1), Chiral purity: 97.4%.
Stage-III-Esterification with methanol in presence of chloromethylsilane
To a well stirred solution of stage-ii (1.0 mole eq) in methanol (5 volumes) at 0°C, charged chlorotrimethylsilane (2.5 mole eq) in methanol (2 volumes) dropwise maintaining 0-5°C and stirred for next 1 hour. The reaction temperature is slowly raised to 25°C and stirred for 7 h. On completion of reaction, as monitored by TLC (MDC: MeOH:: 9:1), methanol is distilled off under vacuum. To the hydrochloride solid obtained, charged MDC (5 volumes) and water (5 volumes) and cooled at 0°C. The pH of the reaction is slowly adjusted to 9.0 using liquid NH3. After settling and separating the layers, organic layer washed twice with water (5 volumes) and dried over sodium sulfate. The MDC layer is distilled under vacuum to obtain yellow oil. Yield: 1 w/w, Mass: 208 (m+1), HPLC purity: 99%.
Stage-IV-Reduction of ester to alcohol in presence of sodium borohydride
In a 4-neck flask, charged NaBH4 (4.0 mole eq) under inert atmosphere in THF (5 volumes). Cooled the reaction mixture to 0°C and stirred for 15 min. To this solution, added dropwise stage-iii (1.0 mole eq) dissolved in THF: methanol::1: 1 (2 volumes each). After the addition, the reaction temperature is raised to 65°C and stirred at this temperature for 9 h. As per TLC monitoring, on the completion of the reaction, the reaction mixture is cooled to 0-5°C. Charged MDC (5 volumes) and water (5 volumes) and extracted the reaction mixture. After settling and separating the layers, organic layer washed twice with water (5 volumes) and dried over sodium sulfate. The MDC layer is distilled under vacuum to obtain yellow oil. Yield: .73 w/w, Mass: 180 (m+1), HPLC purity: 85.06%.
Stage-V-Debenzylation
To a solution of stage-iv and methanol, added 10% of palladium over carbon. This reaction is carried out in an autoclave at 60°C and hydrogenated at 5 kgs pressure. The reaction completion is monitored by TLC. Pd/C is filtered over celite under nitrogen. The filtrate is distilled under vacuum to obtain yellow oil i.e. (R)-3-amino butanol. Yield: .47 w/w, Mass: 90 (m+1), HPLC purity: 98% (after fractional distillation), Chiral purity: 88%.
Conclusion
A simple, efficient, eco-friendly synthetic route is developed involving a biodegradable enzymatic step for the synthesis of (R)-3-amino butanol, highly convenient chemical steps, cost effective procedure. This process is best suitable for the preparation of (R)-3-amino butanol and scalable in the plant.
Acknowledgement
Our group would like to thank the Department of Scientific and Industrial Research India, Dr. Hari Babu (COO Mylan), Sanjeev Sethi (Chief Scientific Office Mylan Inc); Dr Abhijit Deshmukh (Head of Global OSD Scientific Affairs) ; Dr Yasir Rawjee {Head - Global API}, Dr Sureshbabu Jayachandra (Head of Chemical Research); Dr. Suryanarayana Mulukutla (Head Analytical Dept MLL API R & D) as well as analytical development team of Mylan Laboratories Limited for their encouragement and support. We would also like to thank Dr Narahari Ambati (AGC- India IP) & his Intellectual property team for their support.
References
- Z. Temesgen, D.S. Siraj, Ther. Clin. Risk. Manag., 2008, 4(2), 493-500.
- D.E. Dow, J.A. Bartlett, Infect. Dis. Ther., 2014, 3(2), 83-102.
- Process for the preparation of Dolutegravir, 2017, US9573965B2.
- Method for preparing optically pure 3-aminobutyl alcohol, 2012, CN101417954B.
- D. Peña 1, A.J. Minnaard, J.G. De Vries, B.L. Feringa, J. Am. Chem. Soc., 2002, 124(49), 14552-14553.
- B. Weiner, A. Baeza, T. Jerphagnon, B.L. Feringa, J. Am. Chem. Soc., 2009, 131(27), 9473-9474.
- M.R. Saidi, R.S. Brown, F. Rajabi, J. the Iranian Chemical Society., 2005, 4(2), 300-304.
- M. Kazuhiko, Z. Xiaoyong, H. Kiyoto, M. Toshiyuki, O. Tadamasa, S. Hideo, S. Takao, S. Noboru, Org. Process Res. Dev., 2011, 15 (5), 1130-1137.



