Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 6
New Approaches of 4-Aryl-2-Hydrazinothiazole Derivatives Synthesis, Molecular Docking, and Biological Evaluations
Magda H Abdellattif1*, Mostafa A Hussien2,3 and Eman Alzahrani4
1Pharmaceutical Chemistry Department, Deanship of Scientific Research, Taif University, 21974, KSA
2Chemistry Department, Faculty of Science, King Abdulaziz University, KSA
3Chemistry Department, Faculty of Science, Port Said University, Egypt
4Chemistry Department, Faculty of Science, Taif University, 21974, KSA
- *Corresponding Author:
- Magda H Abdellattif
Pharmaceutical Chemistry Department
Deanship of Scientific Research
Taif University, 21974, KSA
Abstract
In the field of the preparation of chemotherapeutic agents with new mechanism distinct from currently approved drugs is important. The implication of different members of Histone Acetyl Transferase (HAT) in cancer disease is well documented in literature, however the clinically effective approved small inhibitor molecule is still lacking. Previous reports indicated that 4-aryl-2-hydrazinothiazole derivatives provided a useful start point to develop HAT inhibitors. Consequently, preparation and biological evaluation of a focused library of 4-aryl-2- hydrazinothiazole based derivatives as useful as anti-cancer agents. Synthesis of 4-aryl-2-hydrazinothiazoles (1a-d), (2a-d), (3a-c), (4a-b), (5ab), (6a-b) studied by either conventional method and free solvent microwave one pot method, the activity against different cancer cell-lines and the antimicrobial activities against different organisms also studied. It was found that all of the prepared thaizoles derivatives were active toward many cell lines (breast, liver, and prostate) where the more active compounds were 4a, 4b, and 6a. Most of these compounds showed highly activity as antibacterial for gram positive and gram negative. However it showed also antifungal activity. Molecular docking studies guided and proved the biological activities of these novel 4-aryl-2-hydrazinothiazoles. Also Infrared Radiation (IR), Proton Nuclear Magnetic Resonance (1H-NMR), Carbon-13 Nuclear Magnetic Resonance (13C-NMR), Gas chromatography–Mass Spectrometry (GC-MS), and elemental analysis were studied.
Graphical Abstract
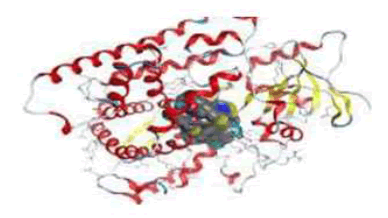
3D plot of the interaction against receptors of 3EQM
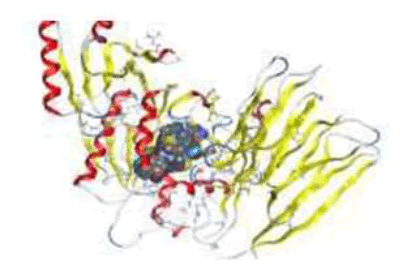
3D plot of the interaction against receptors of 3CQX protein with 4b
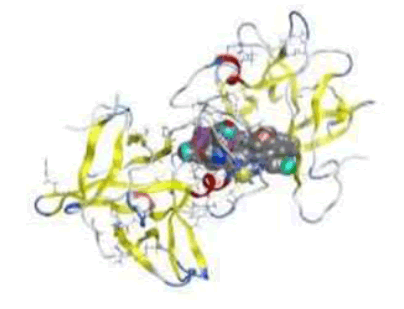
3D plot of the interaction against receptors of 4ihz protein with 4a
Keywords
Antibacterial, Anticancer, Green synthesis, Microwave assisted synthesis, Molecular modeling, 4-Aryl-2-hydrazinothiazol.
Introduction
As the second leading cause of death after cardiovascular diseases, cancer is cumbersome health problem. The ultimate cure of cancer is still lacking. Therefore development of new anticancer agents is needed to satisfy clinical needs. This necessitates enhancing the currently available therapeutic toolbox for combating cancer. This might be achieved by development of novel therapeutic agents based on the less exploited validated targets and thus providing novel anticancer agents with new mechanisms of actions. Apparently, this would also assists in overcoming the emerging resistance to anticancer.
Epigenetic modifications control the compactness of chromatin structure and hence accessibility of DNA genes. Accordingly, expression of genes would be affected by these epigenetic modifications which includes alterations of histones via lysine acetylation, lysine and arginine methylation, serine and threonine phosphorylation, and lysine ubiquitination and sumoylation. The process of acetylation of histones is controlled by writers (Histone Acetyl Transferases; HAT) and erasers (Histone Deacetylases; HDAC). While considerable efforts have been directed to HDAC inhibitors as anticancers, few reports are found for development of HAT inhibitors [1]. Based on homology, HATs are classified into several families. One of these families, four are the most studied and understood which are the GNAT family; the p300/CBP family; the MYST family and the Rtt109 family [2]. The role of two members of the GNAT family, namely PCAF and GCN5, is well documented in cancer [3]. In addition, it has been reported that GCN5 plays an important role in cell cycle progression and transcription of the gene of epidermal growth factor [4,5]. Accordingly, inhibition of GCN5 would lead to antiproliferative effect. Furthermore, another two members of HAT, namely p300 and CBP were reported to have important roles in cells’ G1/S transition [6]. Also, down regulation of p300 was found to result in antiproliferative activity in human melanocytes [7]. Consequently, development of histone acetyl transferase inhibitors could result in molecules with potential anticancer activity.
The 4-Aryl-2-hydarzinothiazole nucleus is a promising scaffold for development of HAT inhibitors. Recently, Cyclopentylidene-[4-(4′- chlorophenyl) thiazol-2-yl) hydrazine 1 was reported [8] as HAT inhibitor. It is commercially available from Sigma-Aldrich Company as a research probe modulating GCN5 member of HAT (CAS Number 357649-93-5).
Opening of the cyclopentane ring to afford 1-(4-(4-chlorophenyl) thiazol-2-yl)-2-(propan-2-ylidene) hydrazine 2 was reported to inhibit GCN5 and p300 members of the HATs and possess antiproliferative activity in neuroblastoma and glioblastoma cell lines [9]. These reports indicate that these structures provide a useful starting point to develop anticancer agents based on HAT inhibition mechanism.
Green chemistry, sustainable synthetic efficiency is becoming important issues in contemporary synthetic practice [10]. Abolishing or minimizing the burden of chemical processes on environmental, ecological systems and limited resources is crucial for survival of forthcoming generations. Replacing organic solvents with water minimize harmful wastes of organic synthesis. In addition, reduction of required reaction time would result in less consumption of energy. Minimizing energy input required to achieve chemical transformations is not only an economical issue, which is of course an important issue, but also is crucial to minimize the environmental burden of power generation plants. Microwave assisted synthesis is a promising tool to achieve these goals of sustainable green synthesis. It enables dramatic reduction of reaction time and hence more efficient energy metrics of the chemical process. Moreover, the microwave assisted synthesis of 2-amino-4-arylthiazoles in aqueous medium has been recently developed [11]. Therefore, it is appealing to develop corresponding strategy for green synthesis of the 4- Aryl-2-hydrazinothiazole nucleus of the proposed research [12-15].
Results and Discussions
Chemistry
1- Synthesis of 4-aryl-2-substitutedthiazoles derivatives
2 - Synthesis of 5-aryl-3-substituted thiazolo[2,3-c][1,2,4]triazoles derivatives
Many attempts were carried out to prepare 1-(4-m-tolylthiazol-2-yl)hydrazine (B) where R = CH3 for derivatives prepared, the first attempt was carried out by conventional method under reflux for 2 h, by refluxing thiocarbohydrazide with, 2-bromo-1-p-tolylethanone in glacial acetic acid [16-19], where the second method was carried out using microwave free reaction for 1.5-2 min. Using the two above compounds without any solvents or liquid media (Schemes 1-5) [20]. The formed compound was confirmed by its Infrared Radiation (IR), Proton Nuclear Magnetic Resonance (1H-NMR), Carbon-13 Nuclear Magnetic Resonance (13C-NMR), and elemental analysis data, the main observation was the presence two broad medium bands one at 600 and the other was at 1400 cm-1, these bands were proved before as a finger print bands as an evidence of the presence of thiazole ring [21]. Not only the time factor or the solvent free factor but also maximum yield product % obtained in comparison to conventional method. The structure of the produced compounds was confirmed by IR, 1H-NMR, 13C-NMR, and elemental analysis, [methodology part]. The main goal of this work was reached by the preparation of different compounds of fused or open structure nitrogen system in a hope to reach different active HAT enzymes (Table 1).
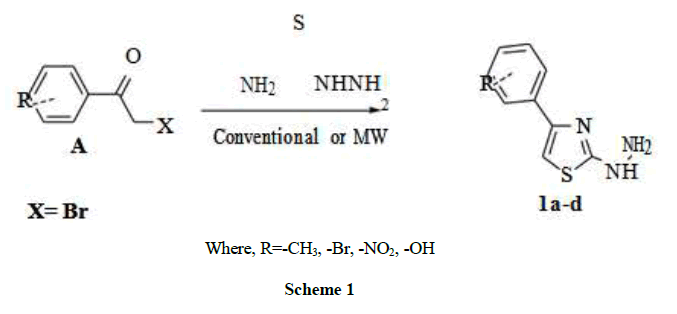
Where, R=-CH3, -Br, -NO2, -OH
Scheme 1
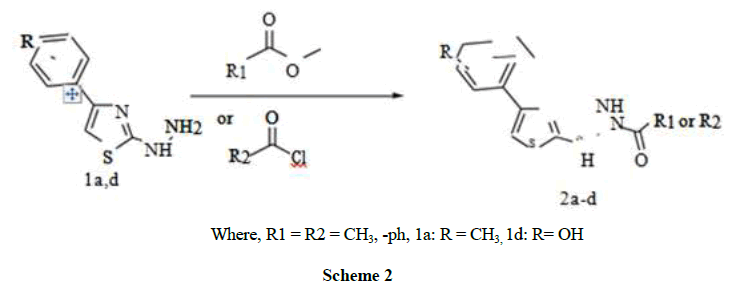
Where, R1 = R2 = CH3, -ph, 1a: R = CH3, 1d: R= OH
Scheme 2
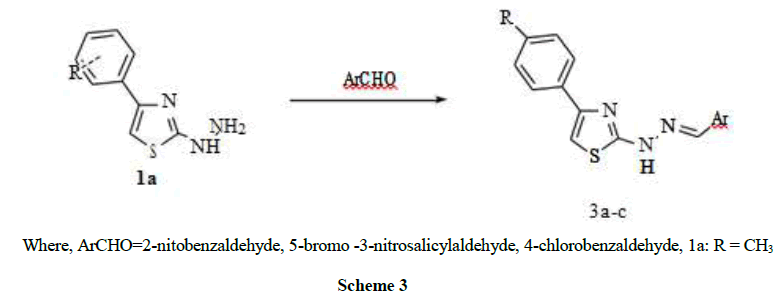
Where, ArCHO=2-nitobenzaldehyde, 5-bromo -3-nitrosalicylaldehyde, 4-chlorobenzaldehyde, 1a: R = CH3
Scheme 3
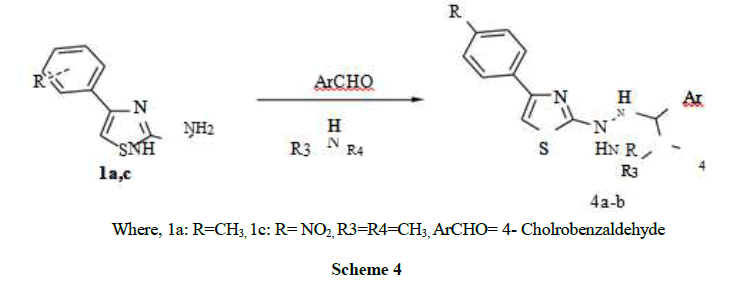
Where, 1a: R=CH3, 1c: R= NO2, R3=R4=CH3, ArCHO= 4- Cholrobenzaldehyde
Scheme 4
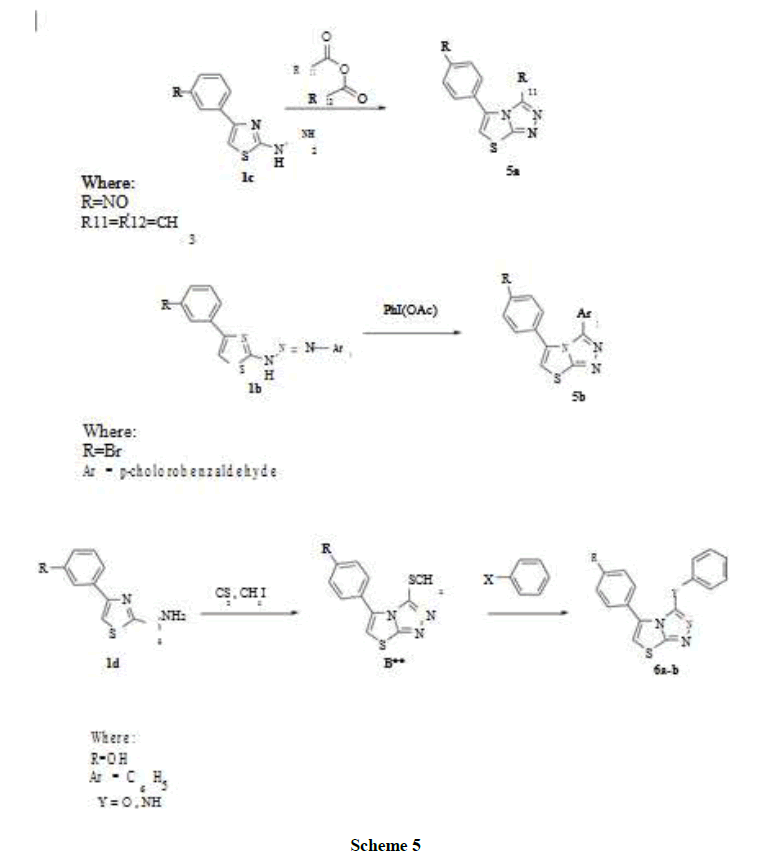
Scheme 5
| Compounds | Name | Structure |
|---|---|---|
| 1a C10H11N3S | [4-(p-Tolyl)-1,3-thiazol-2-yl]hydrazine | 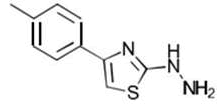 |
| 1b C9H8BrN3S | [4-(p-Bromophenyl)-1,3-thiazol-2-yl]hydrazine | 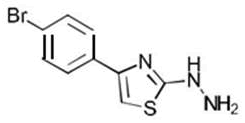 |
| 1c C9H8N4O2S | [4-(p-Nitrophenyl)-1,3-thiazol-2-yl]hydrazine | 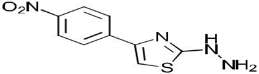 |
| 1d C9H9N3OS | p-(2-Hydrazino-1,3-thiazol-4-yl)phenol | 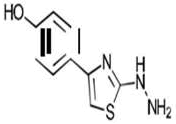 |
| 2a C12H13N3OS | 1-{2-[4-(p-Tolyl)-1,3-thiazol-2-yl]hydrazino}-1-ethanone | 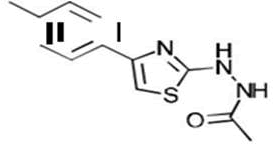 |
| 2b C17H15N3OS | Phenyl{2-[4-(p-tolyl)-1,3-thiazol-2-yl]hydrazino}formaldehyde | 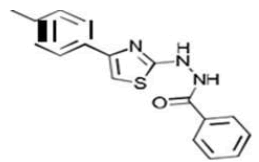 |
| 2c C16H13N3O2S | {2-[4-(p-Hydroxyphenyl)-1,3-thiazol-2-yl]hydrazino}phenylformaldehyde | 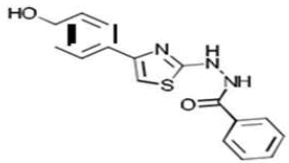 |
| 2d C11H11N3OS | 1-{2-[4-(p-Hydroxyphenyl)-1,3-thiazol-2-yl]hydrazino}-1-ethanone | 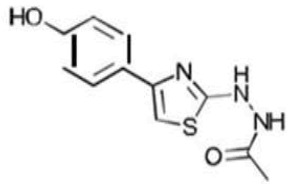 |
| 3a C17H14N4O2S | 2-[(o-Nitrophenyl)methylidene]-1-[4-(p-tolyl)-1,3-thiazol-2-yl]hydrazine | 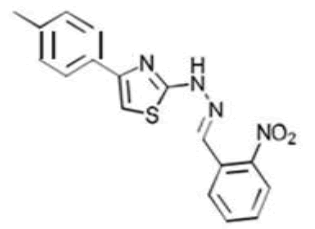 |
| 3b C17H13BrN4O3S | 4-Bromo-2-nitro-6-({2-[4-(p-tolyl)-1,3-thiazol-2-yl]hydrazono}methyl)phenol | 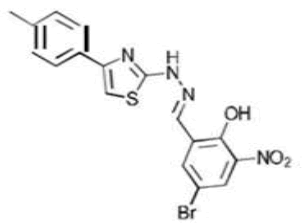 |
| 3c C17H14ClN3S | 1-[(p-Chlorophenyl)methylidene]-2-[4-(p-tolyl)-1,3-thiazol-2-yl]hydrazine | 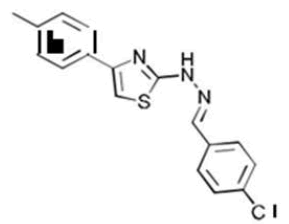 |
| 4a C19H21ClN4S | (p-Chlorophenyl)(dimethylamino){2-[4-(p-tolyl)-1,3-thiazol-2-yl]hydrazino}methane | 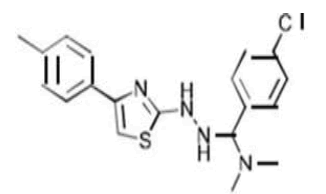 |
| 4b C18H18ClN5O2S | (p-Chlorophenyl)(dimethylamino){2-[4-(p-nitrophenyl)-1,3-thiazol-2-yl]hydrazino}methane | 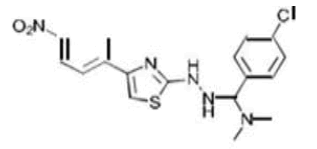 |
| 5a C11H8BrN3S | 8-(p-Bromophenyl)-2-methyl-6-thia-1.3.4-triazabicyclo[3.3.0]octa-2,4,7-triene | 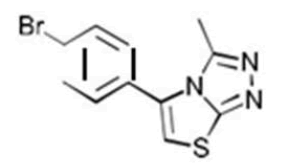 |
| 5b C16H9ClN4O2S | 2-(p-Chlorophenyl)-8-(p-nitrophenyl)-6-thia-1.3.4-triazabicyclo[3.3.0]octa-2,4,7-triene | 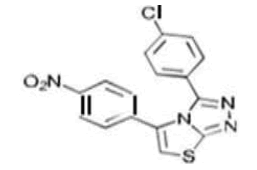 |
| 6a C16H11N3O2S | p-(2-Phenoxy-6-thia-1.3.4-triazabicyclo[3.3.0]octa-2,4,7-trien-8-yl)phenol | 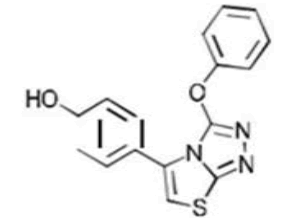 |
| 6b C16H12N4OS | p-(2-Anilino-6-thia-1.3.4-triazabicyclo[3.3.0]octa-2,4,7-trien-8-yl)phenol | 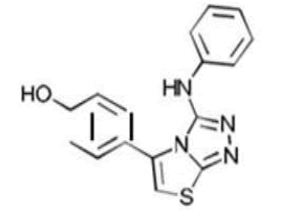 |
| B** C11H9N3S2 | 2-(Methylthio)-8-phenyl-6-thia-1.3.4-triazabicyclo[3.3.0]octa-2,4,7-triene | 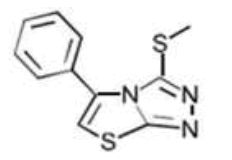 |
Table 1: Structures and Iupac names of the compounds
one could notice from the table above that microwave method was more efficient, although domestic microwave one pot multicomponent was used, the advantages this method are less time consuming and high yield product (Table 2).
| Compound | Conventional method | Microwave method | ||
|---|---|---|---|---|
| Yield % | Time (hrs) | Yield % | Time (min) | |
| 1a | 78 | 02-Mar | 96 | 1.5-2 |
| 1b | 77 | 02-Mar | 94 | 1.5-2 |
| 1c | 72 | 02-Mar | 95 | 1.5-2 |
| 1d | 78 | 02-Mar | 96 | 1.5-2 |
| 2a | 67 | 5 | 94 | 02-Mar |
| 2b | 66 | 04-May | 93 | 02-Mar |
| 2c | 62 | 04-May | 93 | 02-Mar |
| 2d | 67 | 04-May | 92 | 02-Mar |
| 3a | 73 | 04-Jun | 98 | 3-3.5 |
| 3b | 75 | 04-Jun | 97 | 3-3.5 |
| 3c | 71 | 04-Jun | 97 | 3-3.5 |
| 4a | 72 | 8 | 96 | 3.5 |
| 4b | 70 | 8 | 97 | 3.9 |
| 5a | 68 | 12 | 95 | 4 |
| 5b | 74 | 10 | 95 | 3.7 |
| 6a | 70 | 14 | 96 | 2.9 |
| 6b | 70 | 14 | 96 | 3.5 |
| B** | 63 | 9 | 80 | 4.5 |
Table 2: Comparison Yield/time Reaction Using Two Synthetic Techniques
Also most of the compounds obtained were racemic as a result of using mechanical thermal energy of domestic microwave. B** intermediate was separated and characterized also which gave indication and proof that the reactions moved in the right way either by conventional method or by microwave irradiation method. The presence of a short medium band between 600-700 cm-1 indicated the presence of aliphatic C-S bond of - S-CH3.
Biology
Bacteria and fungi had developed resistance strains against currently available antimicrobial agents and therefore it is essential for medicinal chemists to design and synthesize novel antimicrobial agents having less toxicity and more potent effects in much lesser time. In continuation to this, chemists have successfully synthesized effective agents based on heterocyclic compounds. The shining examples are furamizole, nasapadil, tazobactam, and cefatrizine. Thiazoles derivatives have various types of pharmacological effects, including antimicrobial [22].
Numerous examples show that thiazoles and its derivatives play an important role in development of antimicrobial agents. Thiazoles derivatives have been synthesized by the condensation of ophenylenediamine with acids, their nitriles, imidates and orthoesters [23]. It had previously synthesized triazines derivatives and had received some exiting antimicrobial results [24,25]. Therefore, it focused on benzimidazole-based thiazoles derivatives which possess diverse chemical structures. These hybrid structures may be useful for the development of antimicrobial agents. The development of efficient preparation of thiazoles and triazines based compounds played a key role in modern organic synthesis [26].
The minimal inhibitory concentration for some of the newly synthesized compounds showed high significance activity against G-positive and G-negative and antifungal activities. Among the screened compounds, 1b, 1c, 2a, 2b, 3a, 4a, 4a, 5a, and 6a exhibited strong antimicrobial activity [6]. A series of 4-(p-Aryl) -1, 3-thiazol-2-ylhydrazines derivatives are evaluated their antimicrobial activity against Gram positive (Staphylococcus aureus and Strepococcus. pyogenes), Gram negative bacteria (Escherchia coli and Pseudomonas aeruginosa), and strains of fungi (C. albicans, Asperigillus niger, and Asperigillus clavatus), they showed significant antimicrobial activity against tested microorganisms [27].
Antimicrobial
Antibacterial: Table 3 and Figure 1 showed in-vitro experiment for the mean of antimicrobial effects on Gram positive bacteria (SA), that revealed the effect of this dissolved substances as antimicrobial (antibacterial) and were done in the hours of exposures included (1, 3, 5 and 7 h). The mean antimicrobial effects were calculated during the exposure hours and the averages were in descending order by the percentage of the antibacterial as illustrated in Table 3 and Figure 1. Bacteria had developed resistance strains against currently available antimicrobial agents, synthesize novel antimicrobial less toxicity and more potent effects in much lesser time. The shining examples are furamizole, nasapadil, tazobactam, and cefatrizine. Benzimidazoles have various types of pharmacological effects, including antimicrobial [22-26]. The minimal inhibitory concentration for some of the newly synthesized compounds showed high significant activity against Gram positive. Among the screened compounds, the thiazoles derivatives 1b, 1c, 2a, 2b, 3a, 4a, 4a, 5a, and 6a exhibited strong antimicrobial activity [25-27].
| Compound | 1h | 3h | 5h | 7h | Mean |
|---|---|---|---|---|---|
| 1b | 28 | 41 | 61 | 80 | 54 |
| 1c | 32 | 46 | 72 | 100 | 62.5 |
| 2a | 34 | 56 | 84 | 100 | 68.5 |
| 2b | 48 | 71 | 100 | 100 | 79.6 |
| 3a | 31 | 59 | 78 | 100 | 67 |
| 4a | 38 | 62 | 86 | 100 | 71.5 |
| 4a | 42 | 73 | 100 | 100 | 78.8 |
| 5a | 46 | 79 | 100 | 100% | 81.5 |
| 6a | 57 | 83 | 98 | 100 | 84.5 |
Table 3: In-vitro experiment for the mean of antimicrobial effects on G-positive bacteria
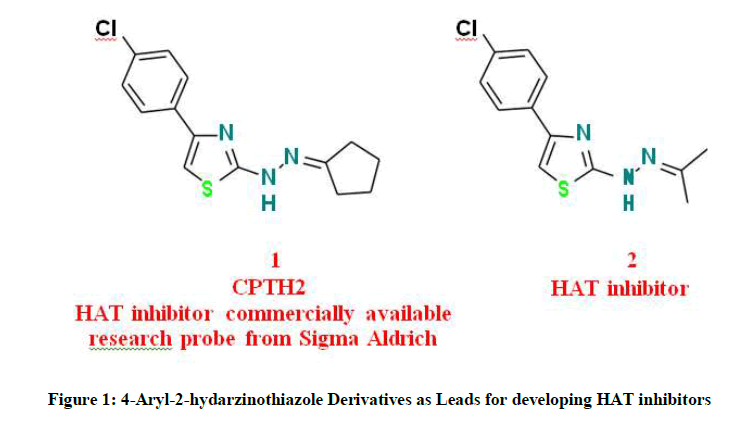
Figure 1: 4-Aryl-2-hydarzinothiazole Derivatives as Leads for developing HAT inhibitors
Table 4 and Figure 2 showed in-vitro experiment for the mean of antimicrobial effects on G-Negative bacteria (EC), that revealed the effect of this dissolved substances as antimicrobial (antibacterial) and were done in the hours of exposures included (1, 3, 5 and 7 h). The mean antibacterial effects were calculated during the exposure hours and the averages were in descending order by the percentage of the antibacterial as shown in Table 4 and Figure 2. The minimal inhibitory concentration for some of the newly synthesized compounds showed high significant activity against G-negative bacteria [26-27].
| Compound | 1h | 3h | 5h | 7h | Mean |
|---|---|---|---|---|---|
| 1b | 27 | 38 | 61 | 86 | 53 |
| 1c | 29 | 40 | 76 | 100 | 61.3 |
| 2a | 30 | 51 | 80 | 100 | 65.3 |
| 2b | 41 | 68 | 82 | 100 | 72.8 |
| 3a | 28 | 51 | 79 | 100 | 64.5 |
| 4a | 32 | 53 | 83 | 100 | 67 |
| 4a | 38 | 67 | 89 | 100 | 73.5 |
| 5a | 40 | 70 | 93 | 100 | 78 |
| 6a | 46 | 78 | 95 | 100 | 81 |
Table 4: In-vitro experiment for the mean of antimicrobial effects on G-Negative bacteria % (*EC)
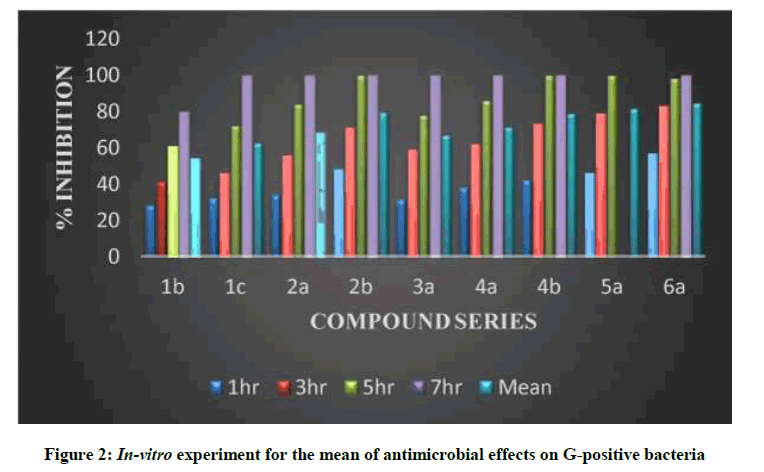
Figure 2: In-vitro experiment for the mean of antimicrobial effects on G-positive bacteria
Antifungal
Table 5 and Figure 3 showed in-vitro experiment for the mean of antimicrobial effects on (CA), that revealed the effect of this dissolved substances as antimicrobial antifungal and were done in the hours of exposures included (1, 3, 5 and 7 h). The mean antifungal effects were calculated during the exposure hours and the averages were in descending order by the percentage of the antifungal as shown in Table 5 and Figure 3. The minimal inhibitory concentration for some of the newly synthesized compounds showed high significant activity against fungi activities [26], their antimicrobial activity fungi (C. albicans) showed significant antimicrobial activity against tested microorganisms [28].
| Compound | 1h | 3h | 5h | 7h | Mean |
|---|---|---|---|---|---|
| 1b | 23 | 33 | 54 | 100 | 52.5 |
| 1c | 24 | 37 | 60 | 89 | 52.5 |
| 2a | 22 | 41 | 69 | 95 | 56.8 |
| 2b | 31 | 48 | 79 | 100 | 64.5 |
| 3a | 23 | 51 | 71 | 96 | 60.3 |
| 4a | 25 | 40 | 59 | 83 | 51.8 |
| 4a | 29 | 58 | 83 | 98 | 67.5 |
| 5a | 39 | 64 | 89.5 | 100 | 75 |
| 6a | 43 | 70.5 | 93.5 | 100 | 84.5 |
Table 5: In-vitro experiment for the mean of antimicrobial effects on fungi (*CA) %
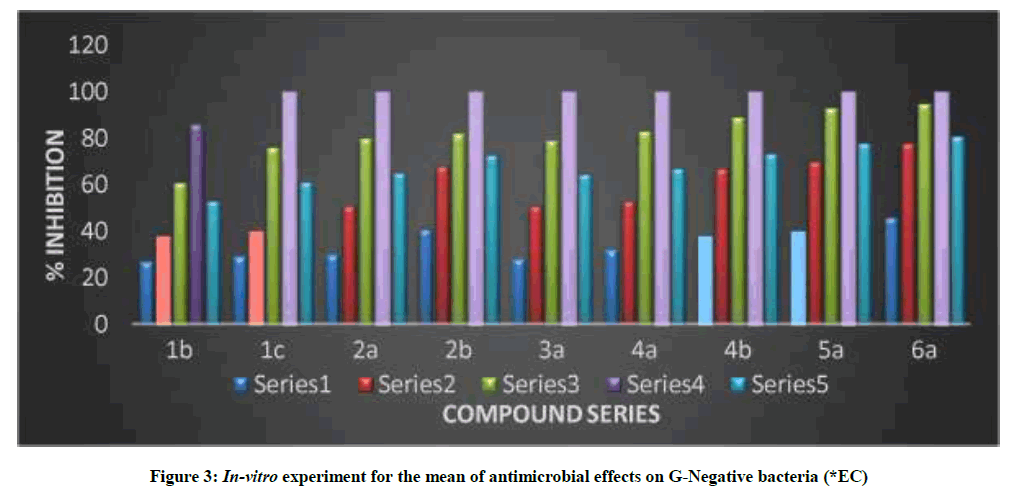
Figure 3: In-vitro experiment for the mean of antimicrobial effects on G-Negative bacteria (*EC)
Anticancer
On the basis of computer modelling technique and the differences in structures of the compounds. As mentioned in literature, many attempts to get HAT inhibitor, New series of 4-Aryl-2-hydrazinothiazole containing nucleus were prepared and investigated toward different thin layers, selected series of the synthetic thiazoles 1b, 1c, 2a, 2b, 3a, 4a, 4a, 5a and 6a were tested in vitro anti-cancer assays against three thin lines of breast cancer, liver cancer, and prostate cancer at constant concentration of 1*10-5 M for each compound and the cultured were incubated for 48 h. Then percentage of inhibition of cell growth was compared with untreated control cells.
The percentage of inhibition for each cell line was illustrated in Table 6, it was found that all of them could inhibit the cell growth by varied extents, but in fact the most effective compounds were 5a, 6a.
| Compound | MCF-7 (breas tcancer) |
HEPG-2 (liver cancer) |
Prostate Cancer (PC-3) |
|---|---|---|---|
| 1b | 44.62 | 62.02 | 43.2 |
| 1c | 33.43 | 59.21 | 65.32 |
| 2a | 24.42 | 53.06 | 61.9 |
| 2b | 48.56 | 33.6 | 67.35 |
| 3a | 49.63 | 57.64 | 81.42 |
| 4a | 51.09 | 61.04 | 88.1 |
| 4a | 56 | 63 | 87.09 |
| 5a | 48.5 | 66.8 | 90 |
| 6a | 47.55 | 47.81 | 84.3 |
Table 6: In vitro anti-cancer activity inhibition %
The novel synthesized compounds were tested for their antitumor activity against three cell lines, MCF-7 (breast cancer), PC-3 (prostate) and HEPG-2 (liver cancer). Compounds 4a, 5a showed the most potent activity, followed by compound 6a; 3b showed the lowest activity (Table 7). These results are in agreement with those obtained by previous researchers [29-32]. These researchers reported that 4-aryl thiazole derivatives had anticancer activity both in vivo and in vitro. From Table 6 and Figure 4 it was found that the most effective compounds for prostate cell line was 5a, 6a, while for 4a was the most effective for hepatic cell line. All of the compounds are active and cytotoxicity of the target compounds were illustrated in Table 7 and Figure 5, IC50.
| Compound | MCF-7 (breast cancer) | HEPG-2 (liver cancer) | Prostate Cancer (PC-3) |
|---|---|---|---|
| IC50 (ug/ml) | |||
| 1b | 35.5 | 34.02 | 35.26 |
| 1c | 20.72 | 25.46 | 25.23 |
| 2a | 31.23 | 32.76 | 21.7 |
| 2b | 35.5 | 29.8 | 27.53 |
| 3a | 29.02 | 37.15 | 18.42 |
| 4a | 21.5 | 13.87 | 14.1 |
| 4a | 21.72 | 13.4 | 11.6 |
| 5a | 26.8 | 19.9 | 19.4 |
| 6a | 27.01 | 23.8 | 24.06 |
Table 7: In vitro anticancer activity 4- against MCF-7 (breast cancer) HEPG-2(liver cancer) and PC-3 (prostate cancer)
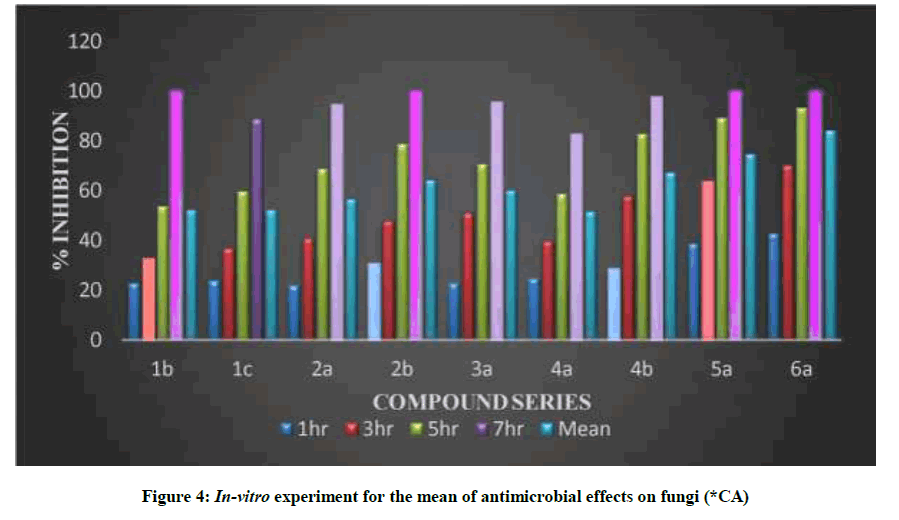
Figure 4: In-vitro experiment for the mean of antimicrobial effects on fungi (*CA)
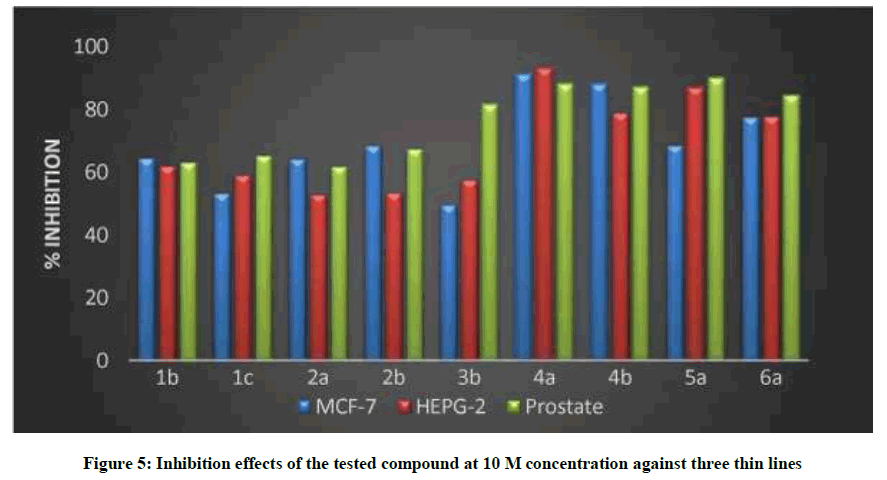
Figure 5: Inhibition effects of the tested compound at 10 M concentration against three thin lines
Molecular Docking Modeling
Molecular modeling studies were performed using Molecular Operating Environment (MOE, 2016) software on an Intel Core i3 processor 1.9 GHz, 4 GB memory with Windows 10, 64-bit operating system (Figure 6). Energy minimizations were performed with RMSD gradient of 0.05 kcal/ mol and MMFF94X force field using MOE and partial charges were automatically calculated. The X-ray crystallographic structures were obtained from Protein Data Bank (PDB), the PDB files are 3eqm, 3cqx, and 4ihz [33]. The target enzymes were prepared for docking by:
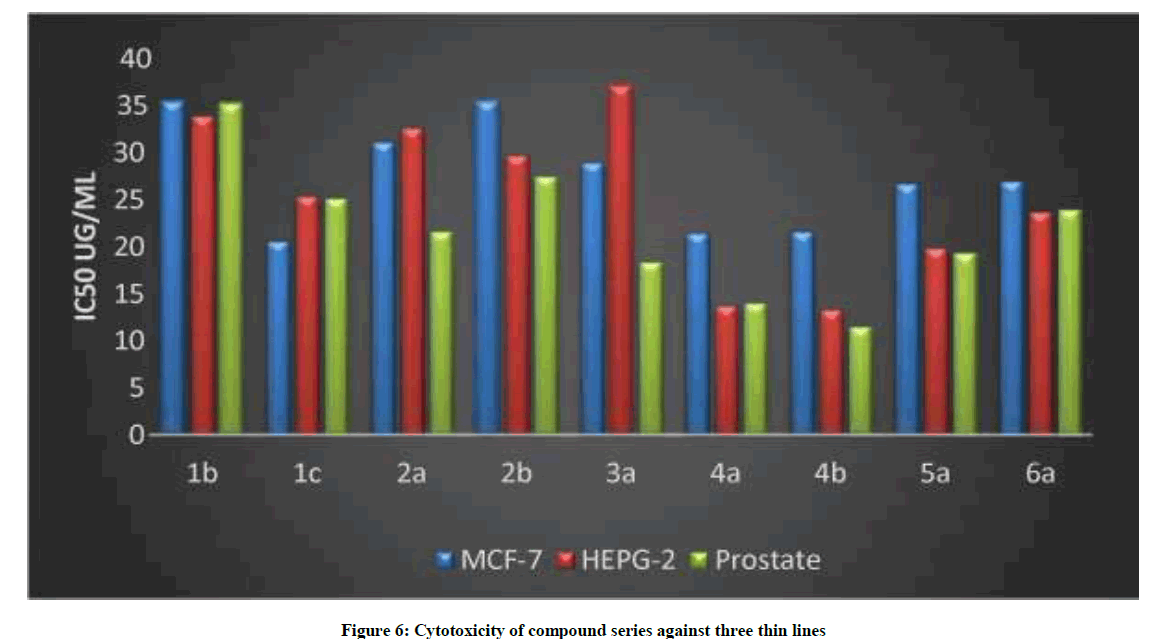
Figure 6: Cytotoxicity of compound series against three thin lines
(i) Removing the ligand from the active site of the enzyme.
(ii) Addition of hydrogen atoms to the structure with their standard geometry.
(iii) Detecting the active site in the enzyme by MOE Alpha Site Finder.
(iv) The obtained pocket was saved as Moe to be used in predicting the ligand enzyme nteractions at the active site and docking of the compounds
A-3EQM: Oxidoreductase / Breast Cancer:
Where, S: final score, which is the score of the last stage That was not set to any; Rmsd: The root mean square deviation of the pose, in Å, from the original ligand. This field is present if the site definition was identical to the ligand definition; rmsd_refine: The root mean square deviation between the pose before refinement and the pose after refinement; E_conf: The energy of the conformer. If there is a refinement stage, this is the energy calculated at the end of the refinement. Note that for Forcefield refinement, by default, this energy is calculated with the solvation option set to Born; E_place: Score from the placement stage; E_score1: Score from rescoring stages 1 and 2; E_refine: Score from the refinement stage, calculated to be the sum of the van der Waals electrostatics and solvation energies, under the Generalized Born solvation model (GB/VI).
From analysis of docked structures it was concluded that the binding models of compound 1b, 1c, 2b, 6a, 3b, 4a, 4b, 6b and X to the crystal structure of 3EQM prostate protein cancer molecule and from the results showed in Table 8, it was found that all of the tested compounds were interacted with the protein through a hydrogen bond, ionic and metal interaction. The values of interaction energies revealed that compounds 4b, 4a and 3b have the most stable interaction than the other complexes and free ligands with minimum binding energy of -9.06 to -8.15 K cal mol-1 see Figure 7.
| Mol | S | Msd | rmsd_refine | E_conf | E_place | E_score1 | E_refine | E_score2 |
|---|---|---|---|---|---|---|---|---|
| 1b | -5.81 | 8.11 | 1.08 | 41.41 | -79.14 | -8.56 | -28.01 | -5.81 |
| 1c | -6.29 | 7.94 | 3.5 | 27.81 | -64.83 | -9.58 | -31.89 | -6.29 |
| 2b | -7.22 | 7.49 | 2.37 | 30.78 | -80.88 | -10.72 | -38.42 | -7.22 |
| 6a | -7.64 | 5.98 | 0.61 | 41.69 | -93.99 | -9.64 | -38.69 | -7.64 |
| 3b | -8.15 | 8.5 | 1.49 | 67.49 | -61.74 | -10.7 | -25.87 | -8.15 |
| 4a | -8.53 | 9.35 | 2.25 | 89.88 | -87.33 | -10.88 | -47.18 | -8.53 |
| 4b | -9.06 | 7.53 | 1.56 | 58.18 | -78.72 | -11.04 | -43 | -9.06 |
| 6b | -7.05 | 8.53 | 0.85 | 45.19 | -78.51 | -9.97 | -28.45 | -7.05 |
| X | -7.34 | 7.11 | 1.59 | 91.74 | -102.96 | -10.42 | -40.7 | -7.34 |
Table 8: Oxidoreductase/3EQM: Oxidoreductase interactions
Figure 7: The Different Binding Modes of 1b, 1c, 2b, 6a, 3b, 4a, 4b, 6b and X with 3EQM protein
3CQX: Protein binding liver cancer:
From analysis of docked structures it was concluded that the binding models of compound 1b, 1c, 2b, 6a, 3b, 4a, 4b, 6b and X to the crystal structure of 3CQX prostate protein cancer molecule and from the results showed in Table 9, it was found that all of the tested compounds were interacted with the protein through a hydrogen bond, ionic and metal interaction. The values of interaction energies revealed that compounds 4b, 4a and 3b have the most stable interaction than the other complexes and free ligands with minimum binding energy of -7.20 to -6.93 K cal mol-1 see Figure 8.
Figure 8: The different binding modes of 1b, 1c, 2b, 6a, 3b, 4a, 4b, 6b and X with 3CQX protein
| Mol | S | Rmsd | rmsd_refine | E_conf | E_place | E_score 1 | E_refine | E_score 2 |
|---|---|---|---|---|---|---|---|---|
| 1b | -5.32 | 13.24 | 2.02 | 42.47 | -31.86 | -9.04 | -26.49 | -5.32 |
| 1c | -5.56 | 15 | 2.74 | 71.69 | -64.64 | -9.56 | -32.4 | -5.56 |
| 2b | -6.35 | 12.61 | 1.62 | 32.36 | -65.18 | -10.97 | -32.02 | -6.35 |
| 6a | -6.42 | 12.69 | 1.36 | 43.38 | -50.94 | -9.2 | -27.96 | -6.42 |
| 3b | -6.93 | 13.02 | 1.1 | 52.79 | -61.24 | -10.18 | -37.45 | -6.93 |
| 4a | -7.04 | 12.06 | 1.33 | 87.86 | -62.76 | -10.65 | -40.07 | -7.04 |
| 4b | -7.2 | 13.59 | 1.37 | 48.15 | -53.9 | -10.12 | -37.7 | -7.2 |
| 6b | -5.79 | 12.06 | 1.28 | 44.95 | -51.82 | -8.91 | -27.88 | -5.79 |
| X | -6.05 | 12.7 | 1.18 | 92.33 | -48.54 | -8.19 | -30.51 | -6.05 |
4IHZ: Hydrolase Inhibitor:
Key of interactions of proteins and molecules
From analysis of docked structures it was concluded that the binding models of compound 1b, 1c, 2b, 6a, 3b, 4a, 4b, 6b and X to the crystal structure of 4ihz prostate protein cancer molecule and from the results showed in Table 10, it was found that all of the tested compounds were interacted with the protein through a hydrogen bond, ionic and metal interaction. The values of interaction energies revealed that compounds 4a, 4b and 3b have the most stable interaction than the other complexes and free ligands with minimum binding energy of -6.62 to -6.25 K cal mol-1 see Figure 9. It concludes that all compounds have good agreement of Molecular docking data with their Biological and anti-cancer Activity showing that Compounds 4a and 4b have the most Biological and anti-cancer activity as shown in Table 11.
Figure 9: The Different Binding Modes of 1b, 1c, 2b, 6a, 3b, 4a, 4b, 6b and X with 4ihz protein
| Mol | S | rmsd | rmsd_refine | E_conf | E_place | E_score1 | E_refine 1 | E_score 2 |
|---|---|---|---|---|---|---|---|---|
| 1b | -4.61 | 8.77 | 3.49 | 40.32 | -25.97 | -7.2 | -20.8 | -4.61 |
| 1c | -4.45 | 11.83 | 5.85 | 69.54 | -3.53 | -7.61 | -19.88 | -4.45 |
| 2b | -5.26 | 9.55 | 2.12 | 31.43 | -9.94 | -5.85 | -21.81 | -5.26 |
| 6a | -5.49 | 12.8 | 2.36 | 42.81 | 22.18 | -5.23 | -25.54 | -5.49 |
| 3b | -6.25 | 12.18 | 2.18 | 50.61 | -16.26 | -7.07 | -29.36 | -6.25 |
| 4a | -6.62 | 8.69 | 1.94 | 84.38 | 48.13 | -7.88 | -28.63 | -6.62 |
| 4b | -6.61 | 12.54 | 1.93 | 52.16 | 9.43 | -8.06 | -31.74 | -6.61 |
| 6b | -5.02 | 10.94 | 4.17 | 48.17 | 91.12 | 0.36 | -20.57 | -5.02 |
| X | -5.45 | 12.04 | 5.44 | 94.75 | 142.28 | 4.24 | -24.82 | -5.45 |
| Comp. | (+ve Gram) | (-ve Gram) | Funga i | MCF-7 (breast cancer) |
(liver cancer) | Prostate | Colon Cancer | leukemia | Docking | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MCF-7 | HEPG-2 | Cancer(PC-3) | (HT29) | MOLT-4 | 3eqm | 3cqx | 4ihz | ||||
| 1b | 28% | 27% | 23% | 44.62 | 62.02* | 43.2 | 33.6 | 53.2 | -5.81 | -5.32 | -4.61 |
| 1c | 32% | 29% | 24% | 33.43 | 59.21 | 65.32 | 45.2 | 55.6 | -6.29 | -5.56 | -4.45 |
| 2a | 34% | 30% | 22% | 24.42 | 53.06 | 61.9 | 36.1 | 52.3 | |||
| 6a | 48% | 41% | 31% | 48.56 | 33.6 | 67.35 | 54.1 | 43.2 | -7.64 | -6.42 | -5.26 |
| 3a | 31% | 28% | 23% | 49.63 | 57.64 | 81.42 | 77.3 | 51.9 | |||
| 4a | 38% | 32% | 25% | 51.09 | 61.04 | 88.1 | 65.2 | 58.4 | -8.15 | -6.93 | -5.49 |
| 4b | 42% | 38% | 29% | 56 | 63 | 87.09 | 76.7 | 63.4 | -8.53 | -7.04 | -6.25 |
| 5a | 46% | 40% | 39% | 48.5 | 66.8 | 90 | 66.7 | 46.5 | |||
| 6b | 57% | 46% | 43% | -6.62 | -6.62 | -6.62 | -6.62 | -6.62 | -6.62 | -6.62 | -6.62 |
| X | -7.34 | -6.05 | -6.61 | ||||||||
| 2b | -7.22 | -6.35 | -5.02 | ||||||||
| 3b | -9.06 | -7.2 | -5.45 | ||||||||
* The activity with high impact related to the average have underline format
Table 11: Comparison between molecular docking data and biological and anticancer activity
Methodology
Chemistry
All chemicals and reagents used were from sigma Aldrich analytical grade and they were used without further purifying and microwave synthesis were carried out using domestic Microwave system. Melting points measurements were determined on (Pyrex capillary) Gallenkamp apparatus. Infrared spectra were recorded with a Thermo Nicolet Nexus 470 Fourier Transform Infrared (FTIR) spectrometer in the range 4000- 400 cm−1 using potassium bromide disks. 1H-NMR spectra, 13C-NMR spectra were recorded on Burker AM250 NMR spectrometer using as DMSO-d6 and CDCl3 solvents for the samples; chemical shifts will be recorded in δ (ppm) units, relative to Me4Si as an internal standard. The mass spectra will be recorded on Shimadzu LCMS-QP 800 LC-MS. Thin-layer chromatography (TLC) was carried out on pre-coated Merck silica gel F254 plates. Elemental analysis was obtained using PerkinElmer 2400 II series CHN Analyser. Each compound series was synthesized by two methods as mentioned before method A (Conventional method), and method B (microwave method).
1a: [4-(p-Tolyl)-1,3-thiazol-2-yl]hydrazine: Method A: A mixture of 4'-methylbromoacetophenone (0.01 mol), with thiosemicarbazide (0.01 mol) in glacial acetic acid (2 mmol) was refluxed under stirring for 2-3 h, after that the mixture cooled at room temperature, the precipitate was collected by filtration and give yellowish crystals.
Method B: A mixture of 4'-methylbromoacetophenone (0.01 mol), with thiosemicarbazide (0.01 mol), was dissolved in methylene chloride/methanol (4: 1, 20 ml), then added silica gel of (1.0 g, 200-400 mesh), the solvent was removed by vaporization, the dried crude was transferred into a glass beaker and irradiated for 1.5-2 min in a domestic microwave oven, the product was tested and separated on silica gel column using methylene chloride as eluent.
1a (C10H11N3S): M. p. 301°C; IR, (KBr, ʋ Cm-1), 810 (Ar), 2900 (-CH3), 3450 (NH2), 3300 (NH); 1H-NMR, δ (ppm)=1.8 (s, 3H, CH3), 3.56(3H, NH, NH2), 7.2-7.6 (m, 4H, Ar); 13C-NMR δ(ppm)=19.28, 77.2, 121.1, 127.5, 132.8, 139.6, 151.3, 156.4, MS (m/z): 205.3 (M+); Anal. Calcd: (205.3), C-58.51; H-5.40; N-20.47, Found: C-58.0; H-5.2; N-20.5.
1b: 4-(p-Bromophenyl)-1,3-thiazol-2-yl]hydrazine: Method A: A mixture of 4'-nitro-bromoacetophenone (0.01 mol), with thiosemicarbazide (0.01 mol) in glacial acetic acid (2 mmol) was reflxed under stirring for 2-3 h, after that the mixture cooled at room temperature, the precipitate was collected by filteration and give yellowish to orange crytstals.
Method B: A mixture of 4'-nitro-bromoacetophenone (0.01 mol), with thiosemicarbazide (0.01 mol), was dissolved in methylene chloride/methanol (4: 1, 20 ml), then added silica gel of (1.0 g, 200-400 mesh), the solvent was removed by vaporization, the dried crude was transferred into a glass beaker and irradiated for 1.5-2 min in a domestic microwave oven, the product was tested and separated on silica gel column using methylene chloride as eluent.
1b (C9H8BrN3S): M. p. 340°C; IR (KBr, ʋ Cm-1), 520 (-Br), 3450 (NH2), 3150 (NH), 780 (Ar); 1H-NMR δ (ppm)=3.8 (3H, NH, NH2), 7.2-7.7 (m, 4H, Ar); 13C-NMR δ (ppm)=77.2, 120.6, 122.1, 129.8, 135.6, 138.7, 151.5, 159.4, MS (m/z): 269.5 (M+); Anal. Calcd: (269), C-58.51; H- 5.40; N-20.47, Found: C-58.51; H-5.40; N-20.47.
1c: [4-(p-Nitrophenyl)-1,3-thiazol-2-yl]hydrazine: Method A: A mixture of 4’-bromo-bromoacetophenone (0.01 mol), with thiosemicarbazide (0.01 mol) in glacial acetic acid (2 mmol) was refluxed under stirring for 2-3 h, after that the mixture cooled at room temperature, the precipitate was collected by filtration and give dark orange crystals.
Method B: A mixture of 4’-bromo-bromoacetophenone (0.01 mol), with thiosemicarbazide (0.01 mol), was dissolved in methylene chloride/methanol (4: 1, 20 ml), then added silica gel of (1.0 g, 200-400 mesh), the solvent was removed by vaporization, the dried crude was transferred into a glass beaker and irradiated for 1.5-2 min in a domestic microwave oven, the product was tested and separated on silica gel column using methylene chloride as eluent.
1c (C9H8N4O2S): M. p. 334°C; IR, (KBr, ʋ Cm-1), 810 (Ar), 1530 (NO2), 3450 (NH2), 3300 (NH); 1H-NMR δ (ppm)=3.8(s, 4H, OH, NH, NH2), 6.8 (dd, 2H, Ar), 7.5 (s H, CHS), 7.6 (dd, 2H, Ar); 13C-NMR δ(ppm)=77.8, 116.35, 134.26, 151.3, 154.5, MS (m/z): 206.9 (M+); Anal. Calcd: (207), C-52.16; H-4.38; N-20.27, Found: C-52.1; H-4.4; N-20.3.
1d: p-(2-Hydrazino-1,3-thiazol-4-yl)phenol: Method A: A mixture of 4’-Hyroxy-bromoacetophenone (0.01 mol), with thiosemicarbazide (0.01 mol) in glacial acetic acid (2 mmol) was refluxed under stirring for 2-3h, after that the mixture cooled at room temperature, the precipitate was collected by filtration and give orange crystals.
Method B: A mixture of 4’-Hyroxy-bromoacetophenone (0.01 mol), with thiosemicarbazide (0.01 mol), was dissolved in methylene chloride/methanol (4:1, 20 ml), then added silica gel of (1.0 g, 200-400 mesh), the solvent was removed by vaporization, the dried crude was transferred into a glass beaker and irradiated for 1.5-2 min in a domestic microwave oven, the product was tested and separated on silica gel column using methylene chloride as eluent.
1c (C9H9N3OS): M. p. above 350°C; IR, (KBr, ʋ Cm-1), 3500 (OH), 3400 (NH2), 3310 (NH), 800 (Ar); 1H-NMR δ (ppm)=3.3 (s, 3H: NH, NH2), 6.8 (dd, 2H, Ar), 7.2 (s, H, CHS), 7.5 (dd, 2H, Ar); 13C-NMR δ (ppm)=77.8, 113.4, 121, 128.3, 129.9, 149, 151.5, 156.3, MS (m/z): 236.5 (M+); Anal. Calcd: (236), C-45.75; H-3.41; N-23.71, Found: C-44.8; H-3.3; N-23.71.
2a: 1-{2-[4-(p-Tolyl)-1,3-thiazol-2-yl]hydrazine}-1-ethanone: Method A: A mixture of 1a (0.01 mol), with acetyl chloride (0.01 mol) in glacial acetic acid (30 ml) was refluxed under stirring for 5 h, after that the mixture cooled at room temperature, then poured on crushed ice, the precipitate was collected by filtration and crystallization from ethanol and give orange to brown crystals.
Method B: A mixture of 1a (0.01 mol), with acetyl chloride (0.01 mol) was mixed with methylene chloride/methanol (4: 1, 20 ml), then added silica gel of (1.0 g, 200-400 mesh), the solvent was removed by vaporization, the dried crude was transferred into a glass beaker and irradiated for 2-3 min in a domestic microwave oven, the product was tested and separated on silica gel column using methylene chloride as eluent.
2a (C12H13N3OS): M. p. above 350°C; IR, (KBr, ʋ Cm-1), 3000 (2 CH3), 3300 (NH), 3320 (NH amide), 1650 (C=O amide); 1H-NMR δ (ppm)=1.5 (s, 3H, CH3), 1.77 (s, 3H, CH3), 6.4 (s, 2H, NH), 7.2-7.5 (m, 4H, Ar), 7.6 (s, H, HCS); 13C-NMR δ (ppm)=14.5, 19.8, 77.6, 121, 127.5, 135.9, 138.7, 151.5, 156.4, 172, MS (m/z): 247.7 (M+); Anal. Calcd (248), C-58.28; H-5.30; N-16.99, Found: C-57.60; H-5.00; N-16.8.
2b: Phenyl{2-[4-(p-tolyl)-1,3-thiazol-2-yl]hydrazino}formaldehyde: Method A: A mixture of 1a (0.01 mol), with benzoyl chloride (0.01 mol) in glacial acetic acid (30 ml) was refluxed under stirring for 4-5 h, after that the mixture cooled at room temperature, then poured on crushed ice, the precipitate was collected by filtration and crystallization from ethanol and give orange crystals.
Method B: A mixture of 1a (0.01 mol), with benzoyl chloride (0.01 mol) was mixed with methylene chloride/methanol (4:1, 20 ml), then added silica gel of (1.0 g, 200-400 mesh), the solvent was removed by vaporization, the dried crude was transferred into a glass beaker and irradiated for 2-3 min in a domestic microwave oven, the product was tested and separated on silica gel column using methylene chloride as eluent.
2b (C17H15N3OS): M. p. above 350°C; IR, (KBr, ʋ Cm-1), 2950 (CH3), 3400 (NH), 3350 (NH amide), 1650 (C=O amide); 1H-NMR, δ (ppm)=1.5 (s, 3H, CH3), 6.1 (s, 2NH), 7.2-7.8 (m, 4HAr, 5Hph, H, HCS); 13C-NMR δ (ppm)=19.8, 77.1, 121, 127.5, 128.7, 130, 133.6, 136, 139, 154.5, 156, 170, MS (m/z): 309.5 (M+); Anal. Calcd: (310), C-66.0; H-5.0; N-13.0, Found: C-65.99; H-4.89; N-13.58.
2c: {2-[4-(p-Hydroxyphenyl)-1,3-thiazol-2-yl]hydrazino}phenylformaldehyde: Method A: A mixture of 1d (0.01 mol), with acetyl chloride (0.01 mol) in glacial acetic acid (30 ml) was refluxed under stirring for 4-5 h, after that the mixture cooled at room temperature, then poured on crushed ice, the precipitate was collected by filtration and crystallization from ethanol and give orange to brown crystals.
Method B: A mixture of 1d with acetyl chloride (0.01 mol) was mixed with methylene chloride/methanol (4: 1, 20 ml), then added silica gel of (1.0 g, 200-400 mesh), the solvent was removed by vaporization, the dried crude was transferred into a glass beaker and irradiated for 2-3 min in a domestic microwave oven, the product was tested and separated on silica gel column using methylene chloride as eluent.
2c (C16H13N3O2S): M. p. above°C; IR, (KBr, ʋ Cm-1), 2890 (2CH3), 3300 (NH), 3320 (NH amide), 1650 (C=O amide), 3540 (OH, phenolic); 1H-NMR δ (ppm)=5.8 (s, H, OH, 2NH), 7.1-7.6 (m, 4HAr, 5Hph, 1HCS); 13C-NMR δ (ppm)=77.8, 116.35, 121, 128.8, 133.5, 134.5, 151, 153.6, 156.7, 170, MS (m/z): 311 (M+); Anal. Calcd: (311), C-61.72; H-4.21; N-13.50, Found: C-60.9; H-4.4; N-13.50.
2d: 1-{2-[4-(p-Hydroxyphenyl)-1,3-thiazol-2-yl]hydrazine}-1-ethanone: Method A: A mixture of 1d (0.01 mol), with benzoyl chloride (0.01 mol) in glacial acetic acid (30 ml) was refluxed under stirring for 4-5 h, after that the mixture cooled at room temperature, then poured on crushed ice, the precipitate was collected by filtration and crystallization from ethanol and give orange crystals.
Method B: A mixture of 1d with benzoyl chloride (0.01 mol) was mixed with methylene chloride/methanol (4: 1, 20 ml), then added silica gel of (1.0 g, 200-400 mesh), the solvent was removed by vaporization, the dried crude was transferred into a glass beaker and irradiated for 2-3 min in a domestic microwave oven, the product was tested and separated on silica gel column using methylene chloride as eluent.
2d (C11H11N3OS): M. p. above 350°C; IR, (KBr, ʋ Cm-1), 2950 (2 CH3), 3300 (NH), 3320 (NH amide), 1650 (C=O amide), 3600 (OH, phenolic), 650 (Ar); 1H-NMR, δ (ppm)=1.46 (s, 3H, CH3), 6.09 (s, OH, 2NH), 7.1-7.5 (m, 4H, Ar, H, HCS); 13C-NMR δ (ppm)=14.8, 77.8, 116.4, 121, 128.9, 134.4, 151.1, 153.5, 156.2, 172.1, MS (m/z): 247 (M+); Anal. Calcd: (247), C-53.00; H-4.45; N-16.86, Found: C-52.8; H-4.7; N-17.00.
3a: 2-[(o-Nitrophenyl) methylidene]-1-[4-(p-tolyl)-1,3-thiazol-2-yl]hydrazine: Method A: A mixture of 1a (0.01 mol), with 2-nitro-benzaldehyde (0.01 mol) in chloro acetyl chloride (10 ml) in DMF (15 ml) was reflxed under stirring for 6 h, after that the mixture cooled at room temperature, then poured on crushed ice, the precipitate was collected by filtration and crystallization from suitable proper solvent.
Method B: A mixture of 1a (0.01 mol), with 2-nitrobenzaldehyde (0.01 mol) in chloro acetyl chloride (10 ml) was mixed with methylene chloride/methanol (4: 1, 20 ml), then added silica gel of (1.0 g, 200-400 mesh), the solvent was removed by vaporization, the dried crude was transferred into a glass beaker and irradiated for 2-3 min in a domestic microwave oven, the product was tested and separated on silica gel column using methylene chloride as eluent.
3a (C17H14N4O2S): M. p. above 350°C; IR, (KBr, ʋ Cm-1), 2850 (CH3), 1000 (C=N free), 3100 (NH), 1340 (NO2), 780 (2Ar); 1H-NMR δ (ppm)=1.46 (s, 3H, CH3), 5.09 (s, NH), 6.9-7.7 (m, 4H, Ar, 4HAr`, H, HCS); 13C-NMR δ (ppm)=19.8, 77.8, 112.4, 116.1, 121, 127.3, 129.9, 135, 139.5, 151, 156, MS (m/z): 338.4 (M+); Anal. Calcd: (339), C-60.34; H-4.17; N-16.56, Found: C-60.6; H-4.3; N-16.45.
3b: 4-Bromo-2-nitro-6-({2-[4-(p-tolyl)-1,3-thiazol-2-yl]hydrazono}methyl)phenol: Method A: A mixture of 1a (0.01 mol), with 5-bromo-3- nitrosalicylaldehyde (0.01 mol) in chloroacetyl chloride (10 ml) in DMF (15 ml) was refluxed under stirring for 4 hrs., after that the mixture cooled at room temperature, then poured on crushed ice, the precipitate was collected by filtration and crystallization from suitable proper solvent.
Method B: A mixture of 1a (0.01 mol), with 5-bromo-3-nitrosalicylaldehyde (0.01mol) in chloroacetyl chloride (10 ml) was mixed with methylene chloride/methanol (4: 1, 20 ml), then added silica gel of (1.0 g, 200-400 mesh), the solvent was removed by vaporization, the dried crude was transferred into a glass beaker and irradiated for 2-2.5 min in a domestic microwave oven, the product was tested and separated on silica gel column using methylene chloride as eluent.
3b (C17H13BrN4O3S): M. p. above 350°C; IR, (KBr, ʋ Cm-1), 2900 (CH3), 3330 (NH), 980 (C=N), 3600 (OH), 1525 (NO2), 570 (Br), 800 (2Ar); 1H-NMR δ (ppm)=1.46 (s, 3H, CH3), 5.09 (s, OH, NH), 7.1-7.7 (m, 4H, Ar, 1HAr`), 7.5 (s, H, HCS); 13C-NMR δ (ppm)=19.8, 77.8, 112.4, 116.1, 118.3, 121, 127.5, 129.9, 135.9, 137.3, 138.2, 139.5, 151.5, 156.4, MS (m/z): 433.8 (M+); Anal. Calcd: (434), C-47.12; H-3.02; N-12.93, Found: C-46.8; H-3.5; N-12.7.
3c: 1-[(p-Chlorophenyl) methylidene]-2-[4-(p-tolyl)-1,3-thiazol-2-yl]hydrazine: Method A: A mixture of 1a (0.01 mol), with 4-chloro-benz-aldehydealdehyde (0.01mol) in benzoyl chloride (10 ml), in DMF (15 ml) was refluxed under stirring for 4 hrs., after that the mixture cooled at room temperature, then poured on crushed ice, the precipitate was collected by filtration and crystallization from suitable proper solvent.
Method B: A mixture of 1a (0.01 mol), with 4-chlorobenzaldehydealdehyde (0.01 mol) in benzoyl chloride chloride (10 ml), was mixed with methylene chloride/methanol (4: 1, 20 ml), then added silica gel of (1.0 g, 200-400 mesh), the solvent was removed by vaporization, the dried crude was transferred into a glass beaker and irradiated for 2-3 min in a domestic microwave oven, the product was tested and separated on silica gel column using methylene chloride as eluent.
3c (C17H14ClN3S): M. p. above 350°C; IR, (KBr, ʋ Cm-1), 850 (Cl), 2950 (CH3), 980 (C=N), 690 (Ar), 3350 (NH); 1H-NMR, δ (ppm)=1.5(s, 3H, CH3), 5.1(s, H, NH, 7.39 (m, 2H, Ar), 7.5 (m, 2H, Ar`), 7.58(s, H, HSN), 7.6 (m, 2H, Ar`), 7.7 (m, 2H, Ar); 13C-NMR, 19.8, 77.7, 77.72, 77.8, 121.07, 126.66, 127.5, 128.95, 129.85, 134.75, 135.86, 138.66, MS (m/z), 327.1,(M+); Anal. Calcd: (327.83), C-63.00; H-4.01; N-12.86, Found: C-62.28; H-4.3; N-12.82.
4a: (p-Chloro-phenyl)-(dimethyl-amino){2-[4-(p-tolyl)-1,3-thiazol-2yl]hydrazino}-methane: Method A: A mixture of 1a (0.01 mol) with dimethyl-amine (0.01 mol), 4-chloro-benz-aldehyde (0.01 mol) in glacial acetic acid (20 ml) was refluxed for 6-8 h, after that the mixture cooled at room temperature, then poured on crushed ice, the precipitate was collected by filtration and crystallization from suitable proper solvent.
Method B: A mixture of 1a (0.01 mol) with dimethylamine (0.01 mol), 4-chlorobenzaldehyde (0.01 mol) was mixed with methylene chloride/methanol (4: 1, 20 ml), then added silica gel of (1.0 g, 200-400 mesh), the solvent was removed by vaporization, the dried crude was transferred into a glass beaker and irradiated for 2-3 min in a domestic microwave oven, the product was tested and separated on silica gel column using methylene chloride as eluent.
4a: (C19H21ClN4S): M. p. above 350°C; IR, (KBr, ʋ Cm-1), 2900 (CH3), 740 (Cl), 800 (Ar), 3400 (2NH), 1250 (C-N); 1H-NMR δ (ppm)=1.49 (s, 3H, CH3), 1.9 (s, 6H, 2CH3), 3.6 (s, NH), 5.4 (s, H, CH), 7.1-7.7 (m, 4H, Ar, 4HAr`), 7.52 (s, H, HCS); 13C-NMR δ (ppm)=19.8, 36.2, 77.8, 87.1, 121, 126.5, 127.7, 129.9, 134.8, 138.2, 151.5, 156.4, MS (m/z): 372.3 (M+); Anal. Calcd: (372), C-61.2; H-5.7; N-15.00, Found: C-61.19; H-5.68; N-15.02.
4b: (p-Chloro-phenyl) (dimethyl-amino){2-[4-(p-nitro-phenyl)-1,3-thiazol-2-yl]-hydrazino}methane: Method A: A mixture of 1c (0.01 mol) with dimethylamine (0.01 mol), 4-chlorobenzaldehyde (0.01 mol) in glacial acetic acid (20 ml) was refluxed for 6-8 h, after that the mixture cooled at room temperature, then poured on crushed ice, the precipitate was collected by filtration and crystallization from suitable proper solvent.
Method B: A mixture of 1c (0.01 mol) with dimethylamine (0.01 mol), 4-chlorobenzaldehyde (0.01 mol) was mixed with methylene chloride/methanol (4:1, 20 ml), then added silica gel of (1.0 g, 200-400 mesh), the solvent was removed by vaporization, the dried crude was transferred into a glass beaker and irradiated for 2-3 min in a domestic microwave oven, the product was tested and separated on silica gel column using methylene chloride as eluent.
4b (C18H18ClN5O2S): M. p. above 350°C; IR, (KBr, ʋ Cm-1), 1525(NO2), 770 (Cl), 800 (Ar), 3400 (2NH), 1250 (C-N); 1H-NMR δ (ppm)=1.8 (s, 6H, 2CH3), 3.6 (s, NH), 5.14 (s, H, CH), 7.1-7.9 (m, 4H, Ar, 4HAr`), 7.5 (s, H, HCS); 13C-NMR δ (ppm)=19.8, 36.2, 77.8, 87.7, 113.3, 126.6, 128, 129.7, 134.8, 148.2, 151, 156.6, MS (m/z): 403.1, (M+); Anal. Calcd: (403), C-53.6; H-4.5; N-17.5, Found: C-53.53; H-4.49; N-17.34.
5a: 8-(p-Bromophenyl)-2-methyl-6-thia-1.3.4-triazabicyclo[3.3.0]octa-2,4,7-triene: Method A: A mixture of 1c (0.01 mol) with acetic anhydride (0.01 mol), in glacial acetic acid (20 ml) and sod. Acetate was refluxed for 12 h, after that the mixture cooled at room temperature, then poured on crushed ice, the precipitate was collected by filtration and crystallization from suitable proper solvent.
Method B: A mixture of 1c (0.01mol) with acetic anhydride (0.01 mol), acetic acid (20 ml) and sod. acetate was mixed with methylene chloride/methanol (4: 1, 20 ml), then added silica gel of (1.0 g, 200-400mesh), the solvent was removed by vaporization, the dried crude was transferred into a glass beaker and irradiated for 3-4 min in a domestic microwave oven, the product was tested and separated on silica gel column using methylene chloride as eluent.
5a (C11H8BrN3S): M. p. 293.4°C; IR, (KBr, ʋ Cm-1), 2150 (triazene), 2850 (CH3), 650 (Br); 1H-NMR δ (ppm)=1.6 (s, 3H, CH3), 7.6-7.8 (m, 4H, Ar, 4HAr`), 8 (s, H, HCS); 13C-NMR δ (ppm)=19.8, 78, 120.7, 123.9, 129.1, 132.3, 139.6, 157.2, 168, MS (m/z): 294, (M+); Anal. Calcd: (294), C-44.91; H-2.74; N-14.28, Found: C-44.86; H-2.64; N-14.00.
5b:2-(p-Chlorophenyl)-8-(p-nitrophenyl)-6-thia-1.3.4-triazabicyclo[3.3.0]octa-2,4,7-triene: Method A: A mixture of 3c (0.01mol) with phI(AcO)2 (0.01 mol), in glacial acetic acid (20 ml) and sod. acetate was refluxed for 10-12 h, after that the mixture cooled at room temperature, then poured on crushed ice, the precipitate was collected by filtration and crystallization from suitable proper solvent.
Method B: A mixture of 3c (0.01 mol) with phI(AcO) 2 (0.01 mol), and sod. acetate was mixed with methylene chloride/methanol (4:1, 20 ml), then added silica gel of (1.0 g, 200-400 mesh), the solvent was removed by vaporization, the dried crude was transferred into a glass beaker and irradiated for 3-4 min in a domestic microwave oven, the product was tested and separated on silica gel column using methylene chloride as eluent.
5b (C16H9ClN4O2S): M. p. above 350°C; IR, (KBr, ʋ Cm-1), 1525 (NO2), 770 (Cl), 800 (2Ar), 1250 (C-N); 1H-NMR 7.1-7.9 (m, 4H, Ar, 4HAr`), 8.2 (s, H, HCS), δ (ppm)=77.9, 113.4, 126.3, 128.1, 129.5, 129.8, 132.7, 140.7, 141.7, 148.9, 157.6, 167.8, MS (m/z): 356.8 (M+); Anal. Calcd: (357), C-53.9; H-2.6; N-15.66, Found: C-53.86; H-2.54; N-15.70.
6a: p-(2-Phenoxy-6-thia-1.3.4-triazabicyclo [3.3.0]octa-2,4,7-trien-8-yl)phenol: Method A: A mixture of 1d (0.01 mol) with carbondisulfide (0.01 mol), methyl iodide (0.01 mol), in ethanol (15 ml) with KOH (5%, 15 ml)/ THF (10 ml) was refluxed for 14 h, which gave in-situ B** as intermediate which could be separated, where B** refluxed in the reaction medium with phenol, then cooled at room temperature, then poured on crushed ice, the precipitate was collected by filtration and crystallization from suitable proper solvent.
6b: p-(2-Anilino-6-thia-1.3.4-triazabicyclo[3.3.0]octa-2,4,7-trien-8-yl)phenol: Method A: A mixture of 1d (0.01 mol) with carbondisulfide (0.01 mol), methyl iodide (0.01 mol), in ethanol (15 ml) with KOH (5%, 15 ml)/ THF (10 ml) was refluxed for 4 h, which gave in-situ B** as intermediate which could be separated, where B** refluxed in the reaction medium with aniline, then cooled at room temperature, then poured on crushed ice, the precipitate was collected by filtration and crystallization from suitable proper solvent.
Method B: A mixture of 1d (0.01 mol) with carbondisulfide (0.01 mol), methyl iodide (0.01 mol), in ethanol (15 ml) with KOH (alcoholic) (5%, 15 ml) then added silica gel of (1.0 g, 200-400 mesh), the solvent was removed by vaporization, the dried crude was transferred into a glass beaker and irradiated for 3-4.5 min in a domestic microwave oven, the product was tested and separated on silica gel column using methylene chloride as eluent. Which gave in-situ B** as intermediate which could be separated, and investigated, then B** was subjected to direct irradiation with phenol to give 6a or with aniline to give 6b.
6a (C16H11N3O2S): M. p. above 350°C; IR, (KBr, ʋ Cm-1), 800 (2Ar), 1250 (C-N), 2150 (triazene) 1100 (C-O), 3550 (OH); 1H-NMR 4.8 (s, H, OH), 7.2-7.8 (m, 4H, Ar, 4HAr`), 7.9 (s, H, HCS); 13C-NMR δ (ppm)=77.8, 114.1, 116.4, 119.8, 128, 129, 134.3, 153.5, 157.1, 158.1, 160, 168.1, MS (m/z): 309 (M+); Anal. Calcd: (309) C-62.12; H-3.58; N-13.58, Found: C-62.00; H-3.68; N-13.6.
6b (C16H12N4OS): M. p. above 350°C; IR, (KBr, ʋ Cm-1), 800 (2Ar), 1250 (C-N), 2150 (triazene) 1100 (C-O), 3600 (NH), 3500 (OH); 1H-NMR 4.6 (s, H, OH, NH), 6.8-7.7 (m, 4H, Ar, 4HAr`), 7.6 (s, H, HCS); 13C-NMR δ(ppm)=77.8, 114.1, 116.4, 117.8, 128, 129, 134.3, 148.5, 158.1, 160, 169, MS (m/z): 307.6 (M+); Anal. Calcd: (308), C-62.32; H-4.0; N-18.17, Found: C-62.2; H-3.92; N-18.2.
B** (C11H9N3S2): M. p. 255.4°C; IR, (KBr, ʋ Cm-1), 800 (Ar), 630 (C-S), 3000 (CH3); 1H-NMR 1.8 (s, 3H, CH3), 7.56-7.8 (m, 6H, 2HAr, 4HAr`), 8.05 (s, H, HCS); 13C-NMR δ (ppm)=77.9, 126.33, 126.55, 127.45, 129, 131.3, 141.6, 157.22, 167, 8, MS (m/z): 307.6 (M+); Anal. Calcd: (308), C-62.32; H-4.0; N-18.17, Found: C-62.2; H-3.92; N-18.2.
Biology
Antimicrobial
Collected the chemical subject samples under study in sterile and dry Eppendorf tubes with the specific information for each sample. Weight the under study quantity of each sample and were dissolved in Acetone, mixed for (2-5) min. Prepared 2 ml of each Acetone dissolved subject in Wizerman tube for each microbe under test and added the data on the tube [34].
Collected the microbial identified isolates as pure and clear colonies, and then made a microbial distilled water suspension of each as McFarland concentration with special information setting [35]. Applied a mixture of (Acetone dissolved subject + microbial suspension), then mix the ingredients and calculated the time. Cultured got from each mixture every time recorded (1, 3, 5 and 7 h), that did on suitable dish media with data recorded on each plate, incubated at (35-37)ºC for (24-48) h. Followed the results of microbial cultures growth and recorded the number of colonies in each dish [36]. Determined the percentage of microbial death by applying the Equation: [(Colony No/300 X 100) -100] [37]. Statistical analysis of the results, calculation of the arithmetic mean for each chemical materials subject samples under study that were showed in the form of tables and graphs [38].
Anticancer
Cell Culture: Many human Cell lines were obtained from Egyptian Holding Company for Biological Products & Vaccines (VACSERA), Giza, Egypt, and maintained in the tissue culture unit. The cells were routinely grown in RBMI-1640 medium, supplemented with 10% heat-inactivated FBS, 50 units/ml of penicillin and 50 mg/ml of streptomycin and maintained in a humidifiedatmospher containing 5% CO2. They were act as a monolayer culture by serial sub-culturing. The reagents of the cell cultured were obtained from Lonza (Basel, Switzerland). The activity of the compounds under investigation as anticancers was determined against MCF-7 cells (breast cancer), PC-3 (prostate cancer), and HEPG-2 cells (liver cancer) [39].
Cytotoxicity: The sulforhodamine B (SRB) was used to determine Cytotoxicity using assay method as previously described by [40]. Collections of Multitimes growing cells were done by using 0.25% Trypsin EDTA and seeded in 96 well plates at 1000-2000 cells/well in RBMI-1640- supplemented medium. After 24 h, cells were incubated for 72 h with different multiple concentrations of the synthesized under investigation compounds. After 72 h treatments, the cells were fixed with 10% trichloroacetic acid for 1 h at 4°C. Wells were stained for 10 min at room temperature with 0.4% SRBC (sulforhodamine B) dissolved in 1% acetic acid. The plates were subjected to air dried for 24 h, and the dye was solubilized with Tris HCl for 5 min on a shaker at 1600 rpm. OD (the optical density) of each well was measured spectrophotometrically at 564 nm with an ELISA microplate reader (ChroMate, 4300, FL, USA). Calculations of IC50 values were be according to the Boltzmann sigmoidal equation for the response of concentration curve by nonlinear regression fitting models (Graph Pad, Prism Version 5).
Modelling: Molecular docking studies were carried out using the Molecular Operating Environment (MOE) docking software [41,42]. The three dimensional (3D) crystal structure of GPb was downloaded from the Protein Data Bank [43,44]. The allosteric site as characterized in the crystal structure of the selected proteins was used as the target site. Before the docking experiment was performed, the compounds prepared for docking by minimizing its energy at the B3LYP/631G (d) level of theory. Partial charges were calculated by Gasteiger’s method. Since most macromolecular crystal structures contain little or no hydrogen coordinate data due to limited resolution, protonation was done prior to docking using Protonate 3D tools implemented in MOE. Protonation was followed by energy minimization up to a 0.05 Gradient using an Amber 99 force field. The compounds were docked into the protein’s active site using the Triangular Matching docking method and thirty conformations of the title compound and protein complex were generated with a docking score (S). The complexes were analyzed for interactions and their 3D images were taken by using visualizing tool PyMol. The docking protocol predicted the same conformation as was present in the crystal structure with an RMSD value within the allowed range of 2.0 Å [45,46]. The superposition of the docked conformation and co-crystalized ligand is shown. Amongst the docked conformations, the top-ranked conformation was visualized for ligand-protein interactions in PyMol software.
Conclusion
In Brief, Synthesis of thaizoles derivatives by domestic microwave method was more efficient than conventional for many reasons like (time, yield, easy ability, solvents, etc.) and also gives us a guide to prepare many compounds by this way. Many of 4-Aryl-2-hydrazinothiazole derivatives were obtained but actually the open structured derivatives were more efficient as antibacterial and as anticancer agents, this may be due the free active N of (NH2, NH-) in some compounds or free (O) especially for 6 series compounds. Molecular docking modelling for the synthesized compounds directed us to the efficiency power of some compounds which is agreed with anticancer and antimicrobial experiments. Some of compounds showed two active sites at docking results which may be later lead us to other investigations for them.
Acknowledgement
The authors are very grateful to Taif University, Taif, KSA, because this work was financial supported by Taif University, Taif, KSA, under project number 1/438/4938.
References
- J.M. Gajer, S.D. Furdas, A. Gründer, M. Gothwal, U. Heinicke, K. Keller, F. Colland, S. Fulda, H.L. Pahl, I. Fichtner, W. Sippl, M. Jung, Oncogenesis., 2015, 4, e137.
- F.J. Dekker, H.J. Haisma, Drug Discov Today., 2009, 14, 942.
- Z. Nagy, L. Tora, Oncogene., 2007, 26, 5341.
- P. Cheung, K.G. Tanner, W.L. Cheung, P. Sassone-Corsi, J.M. Denu, C.D. Allis, Mol. Cell., 2000, 5, 905.
- L.A. Howe, D. Auston, P. Grant, S. John, R.G. Cook, J.L. Workman, L. Pillus, Genes Dev., 2001, 15, 3144.
- S. Ait-Si-Ali, A. Polesskaya, S. Filleur, R. Ferreira, A. Duquet, P. Robin, A. Vervish, D. Trouche, F. Cabon, A. Harel-Bellan, Oncogene., 2000, 19, 2430.
- D. Bandyopadhyay, N.A. Okan, E. Bales, L. Nascimento, P.A. Cole, E.E. Medrano, Cancer Res., 2002, 62, 6231.
- F. Chimenti, B. Bizzarri, E. Maccioni, D. Secci, A. Bolasco, P. Chimenti, R. Fioravanti, A. Granese, S. Carradori, F. Tosi, P. Ballario, S. Vernarecci, P. Filetici, J. Med. Chem., 2009, 52, 530.
- D. Seccic, S. Carradoric, B. Bizzarric, A. Bolascoc, P. Ballariob, Z. Patramania, P. Fragapanea, S. Vernareccib, C. Canzonettab, P. Fileticia, Bioorg. Med. Chem., 2014, 22, 1680.
- C.J. Li, B.M. Trost, Proc. Green chemistry for chemical synthesis, Natl. Acad. Sci., U.S.A, 2008,105, 13197.
- K.S. Jain, J.B. Bariwal, M.K. Kathiravan, V.K. Raskar, G.S. Wankhede, N.A. Londhe, S.N. Dighe, Green and Sustainable Chemistr., 2011, 1.
- C.O. Kappe, D. Dallinger, Nat. Rev. Drug Discov., 2006,5, 51-64.
- K.S. Suslick, Faraday Discuss.,2014, 170,411-422.
- W.D. Shipe, S.E. Wolkenberg, C.W. Lindsley, Drug Discov. Today Tech., 2005, 2, 155-161.
- N.E. Leadbeater, Chem. Commun., 2005, 2881-2902.
- G.C. Saha, K. Khayer, R. Islam, S.K. Chowdhury, Indian J. Chem., 1992, 31, 547-550.
- V. Varshney, N.N. Mishra, P.K. Shukla, D.P. Sahu, Bioorg. Med. Chem. Lett., 2009,19, 6810-6812.
- A. Dornow, H. Menzel, P. Marx, Chem. Ber., 1964, 97, 2173-2178.
- H.A. El-Sayed, A.H. Moustafa, A.Z. Haikal, E.S.H. El-Ashry, Nucleosides Nucleotides Nucleic Acids.,2009,28, 184-192.
- H.A. Saad, M.M. Youssef, M.A. Mosselhi, Molecules., 2011, 16, 4937-4957.
- S.M. Notley, Langmuir., 2012, 28, 14110-14113.
- B.S. Holla, K.V. Malini, B.S. Rao, B.K. Sarojini, N.S. Kumari, Eur. J. Med. Chem., 2003, 38(3), 313-318.
- U. Sekar, B. Padalkar, V. Gupta, K. Pharangare, V. Patil, P. Umape, Arab. J. Chem., 2016, 9, S1125-S1130.
- S. Alaqeel, J. Saudi Chem. Soc., 2017, 21, 229-237.
- N. Desai, V. Joshi, K. Rajpara, H. Vaghani, H. Satodiya, K. Bhatt, Anti-infective agents., 2014, 12, 85-94.
- N.C. Desai, N.R. Shihory, G.M. Kotadiya, Chin. Chem. Lett., 2014, 25, 305-307.
- A. Padmaja, C. Rajasekhar, A. Muralikrishna, V. Padmavathi, Eur. J. Med. Chem., 2011, 6, 5034-5038.
- R. Haggam, H. El-Sayed, S. Said, M. Ahmed, A. Moustafa, R. Abd-El-Noora, J. Heterocyclic. Chem., 2017, 54, 375-383.
- N. Desai, P. Darshan, V. Darshita, Med. Chem. Res., 2017.
- N. Esmaeili, H. Ebrahimzadeh, K. Abdi, S. Safarian, Pharmacogn. Mag., 2011, 7(25), 74-80.
- S. Steven, L.H. Richard, A.Y. Stephen, D.L. Weidbrauk, Clinical virology manual, 4th (Edn.), ASM press, 2009, 1-692.
- P. Karegoudar, M.S. Karthikeyan, D.J. Prasad, M. Mahalinga, B.S. Holla, N.S. Kumari, Eur. J. Med. Chem., 2008, 43(2), 261-7.
- H.I. El-Subbagh, A.M. Al-Obaid, Eur. J. Med. Chem., 1996, 31(12), 1017-1021.
- M. Walaa, M. Nessma, M. Gehad, J. Org. Chem., 2017, 848, 288-301.
- M.H. Abdellattif, M.M. Helmy, H.A. Eldeab, IJAPBC., 2014, 3(4), 984-990.
- CLSI (Clinical and Laboratory Standards Institute) Performance for antimicrobial disk susceptibility tests; approved standard. 11. Wayne (PA), USA, Clinical and Laboratory Standards Institute, 2012.
- https://archive.org/stream/manualofmicrobio00amer/manualofmicrobio00amer_djvu.txt
- https://arxiv.org/pdf/1701.05870.pdf
- https://www.iarc.fr/en/publications/pdfs-online/epi/cancerepi/CancerEpi-3.pdf
- M. Macchia, S. Barontini, S. Bertini, V. Di Bussolo, S. Fogli, E. Giovannetti, E. Grossi, F. Minutolo, R. Danesi, J. Med. Chem., 2001, 44(23), 3994-4000.
- M.T. Abdel-Aal, Arch. Pharm. Res., 2010, 33(6), 797-805.
- M.A. El-Hashash, S.A. Rizk, A.A. El-Sayed, J. Adv. Chem., 2017, 13(12), 6106-6117.
- F. Hegelund, R.W. Larsen, M.H. palmer, J. Mol. Spectroscopy., 2007, 244(1), 63-78.
- A. Barakat, A.M. Al-Majid, S.M. Soliman, G. Lotfy, H.A. Ghabbour, H.K. Fun, A. Wadood, I. Warad, J.C. Sloop, Molecules., 2015, 20(11), 20642-20658.
- M.H. Abdellattif, I.A. Maghrabi, M.M.H. Areef, H.A. El Deab, S.M. Mouneir, A. Belal, JAC., 2016, 12(4), 4351-4364.
- M.K. Paul, A.K. Mukhopadhyay, Int. J. Med. Sci.,2004,1, 101-115.



