Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 5
Molecular Docking Studies of Cu(II) Complex: A Novel Therapeutic Alternative in Treating Staphylococcus aureus< and Related Complications
Naveen Kumar RM1, Govindaraju Shruthi2, Shiva Prasad Kollur3, Chandan Shivamallu2*, Mekhala N Chitagudigi2 and Prasad N4*
1Vijaya Vittala College, Mysore, Karnataka, India
2Faculty of Life Sciences, Division of Biotechnology & Bioinformatics, JSS Academy of Higher Education, Mysuru, Karnataka-570015, India
3Manipal Centre for Natural Sciences, Manipal Academy of Higher Education, Manipal, Karnataka-576104, India
4Department of Chemistry, Government Engineering College, Chamarajanagar, Karnataka-571313, India
- *Corresponding Author:
- Chandan Shivamallu
Faculty of Life Sciences
Division of Biotechnology & Bioinformatics
JSS Academy of Higher Education
Mysuru, Karnataka-570015, India
Prasad N
Department of Chemistry
Government Engineering College
Chamarajanagar, Karnataka-571313, India
Abstract
Copper is one of the essential metal ions required by Staphylococcus aureus (S. aureus) as a cofactor for synthesis and activity of various enzymes which includes superoxide dismutase. Present work describes the computational studies, particularly, molecular docking investigations of Cu(II) complex in the inhibition of Superoxide Dismutase (SOD) produced in the pathogen S. aureus. The results from this study revealed that the Cu(II) complex will be readily taken up by the organism thereby facilitating easy inhibition or inactivation of SOD. This is due to high affinity of SOD towards copper core of the complex.
Keywords
Cu(II) complex, Staphylococcus aureus, Molecular dockling, Superoxide dismutase
Introduction
Copper(II) coordination compounds act as inhibitors of respiratory dehydrogenases in electron transport chain [1]. Particularly, coordination compounds of Cu(II) ions transport toxic Cu(II) ions into the cytoplasm of the microorganism thereby increasing the antimicrobial activity of copper complexes, these then disrupt the normal functioning of various essential and accessory proteins of bacteria and fungi such as Superoxide Dismutase (SOD) which is involved in converting oxygen free radicals produced during oxidative burst useless. A highly virulent organism, Staphylococcus aureus, exhibit such mechanism in which well-defined SOD protein is found to get entry into the host without much resistance. Also, S. aureus is found to possess high antimicrobial resistance with least or no effect of antibiotic such as penicillin and tetracycline on the organism [2,3]. This calls for an immediate alternative therapy for treating against pathogen without being neutralized by the organism to provide relief to this problem.
In continuation of our previous work [4], extended in the present work to focus on specificity of action of Cu(II) complex against SOD, which is found to be produced exogenously by the host cell by the action of phagocytic cells, contributing to virulence in S. aureus. The ability of Cu(II) complex to inhibit the protein has been studied using in silico molecules docking studies and various bioinformatics tools such as Swiss model workspace, I-Tasser, Argus lab, and Chimera.
Materials and Methods
Homology modelling/ structure prediction of SOD
The protein sequence (FASTA sequence) of SOD of S. aureus was obtained from GenBank (NCBI databases) and the sequence obtained was submitted to three softwares, Swiss model, LOMETS, I-TASSER to obtain structures with precision.
Swiss-model workspace
A homology modelling tool, it screens the entire protein data bank for similarity in the sequence followed by screening the structures in the protein data bank for structural similarities based on secondary structure analysis followed by prediction of highly acceptable structure of the functional protein, with details regarding the GQ mean and also description regarding the function of the protein based on its quaternary structure [5-8].
CPH model 3.2 server
In this server the model is generated by performing iterative building profile by aligning the submitted query sequence to the sequences of non-reductant protein database to obtain a appropriate template required to build the model. The built model represents the quaternary structure of the functional protein [9].
Interactive Threading Assembly Refinement (I-TASSER)
It is a server in which the secondary structure of query sequence is generated, which is matched with the secondary structure of protein database to obtain a template, this is done using LOMETS. This is followed by assembling of PDB templates into full length models through replica exchange Monte Carlo simulations with unaligned regions which includes loops built through ab initio modeling upon non-retrieval of the templates due to lack of matching template, the whole model is built through ab initio modelling using LOMETS and I-TASSER. The low free energy states are identified through clustering the simulation decoys using SPICKER [10-12].
Validation of protein structure
The predicted protein structure was validated through Ramachandran plot prediction for the amino acid residues in the protein. This was done to verify the number of favorable, allowed and disallowed residues in the predicted protein structure of SOD. Rampage software was used to predict the Ramachandran plot for the submitted protein structure of SOD in PDB format [13].
Generation of 3D structure of Cu(II) complex
The structure of Cu(II) complex obtained through Nuclear magnetic resonance (NMR) and Mass spectrometry (MS) in our previously published work (4) was drawn in silico using Chemdraw (Ver.8.0) software. The chemdraw file was then converted to Mol 2 by addition of 3D coordinates to the 2D structure of Cu(II) complex using OpenBabel software. The obtained 3D structure file of the complex molecule was further refined before molecular docking.
Molecular docking
In silico molecular docking of Cu(II) Complex to SOD of S. aureus was done using Argus lab software to analyze the ability of Cu complex to bind to SOD and inhibit its action and also to find the degree of affinity of Cu complex towards the protein binding site. To analyze this binding site of SOD, screening was done using BSpred, Coach and RaptorX servers to eliminate ambiguity and to obtain high precision in prediction of binding site. After prediction of binding site, the protein structure was loaded in Argus lab software and the binding site was defined. Now, the ligand in Mol 2 format was loaded in Argus lab and the structure was cleaned before docking and hydrogens were added, followed by generation of grid for docking. The docking was performed using Argus lab algorithm with flexible docking parameters [11,12,14-21].
Visualization
The docked results were visualized using CHIMERA visualization software, where the interactions between complex and protein binding site was visualized by the bonds between interacting atoms in complex and protein [10,14,22-30].
Results and Discussions
Structure validation report
The Ramachandran plot obtained through Rampage software upon analyzing SOD structure obtained through homology modelling using Swiss-model workspace, I-TASSER, CPH model servers showed 95.9% of the amino acid residues in the favored region, 4.1% residues were found to occur in allowed region with 0% of residues in outlier region (Figure 1).
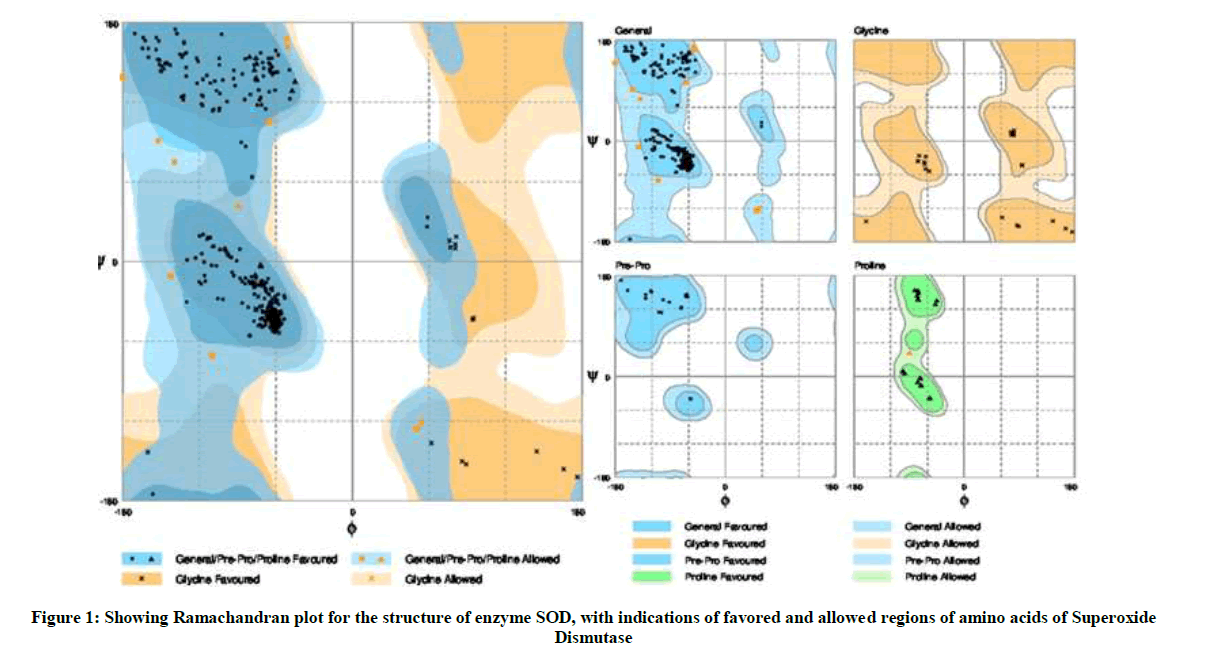
Figure 1: Showing Ramachandran plot for the structure of enzyme SOD, with indications of favored and allowed regions of amino acids of Superoxide Dismutase
Binding site prediction
The binding site prediction by BSpred, Coach and RaptorX servers reported that the amino acids THR 122, LEU 123, PHE 124 and THR 142 form the binding site in SOD of S. aureus (Figure 2).
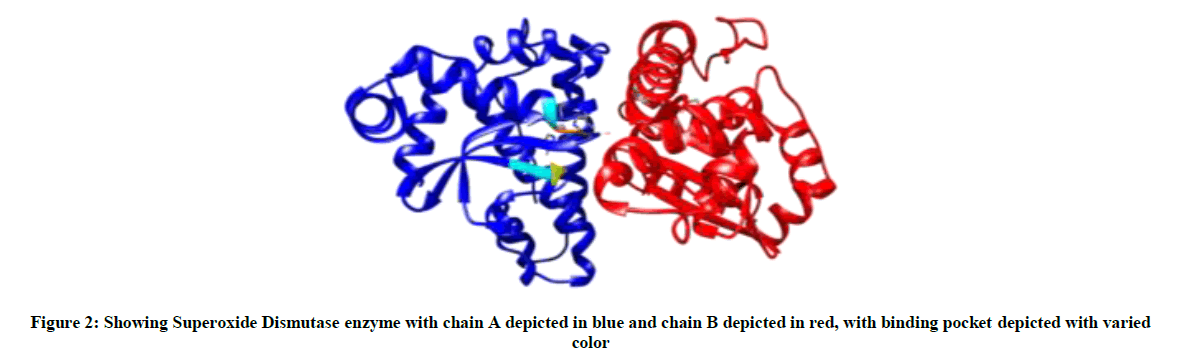
Figure 2: Showing Superoxide Dismutase enzyme with chain A depicted in blue and chain B depicted in red, with binding pocket depicted with varied color
Molecular Docking
Upon obtaining binding site residues of SOD, the residues which constitute the binding site were selected and assigned as binding site in Argus lab software, cleaning and addition of hydrogens resulted in functional ligand structure of Cu(II) complex (Figure 3).
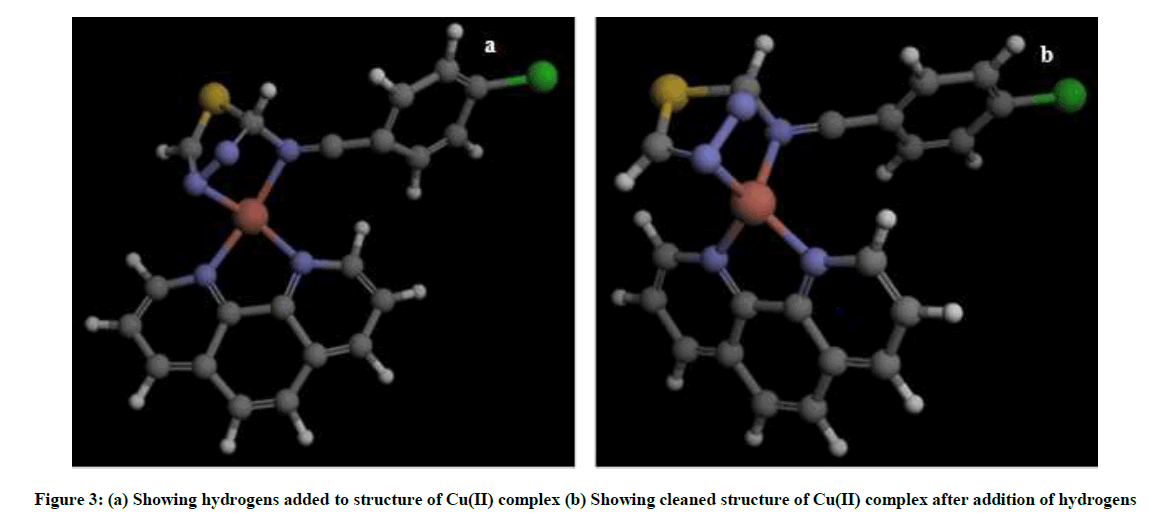
Figure 3: (a) Showing hydrogens added to structure of Cu(II) complex (b) Showing cleaned structure of Cu(II) complex after addition of hydrogens
Molecular docking of Cu(II) complex after grid generation to SOD resulted in 15 best possible docking poses, out of which pose 1 showed least free energy (ΔG) of −7.62 kcal/mol. The Cu(II) complex in pose 1 was found to interact not only with THR 122, LEU 123, PHE 124, THR 142, which form the binding site but also with THR 143, PRO 144, ASN 270, MET 266, ASN 342 residues. Furthermore, the Cu(II) complex binds to the protein SOD at the interphase of chain A and chain B of SOD (Figure 4) with higher interaction than the natural ligands of SOD with lesser free energy, thereby acting as non-competitive inhibitor of SOD.
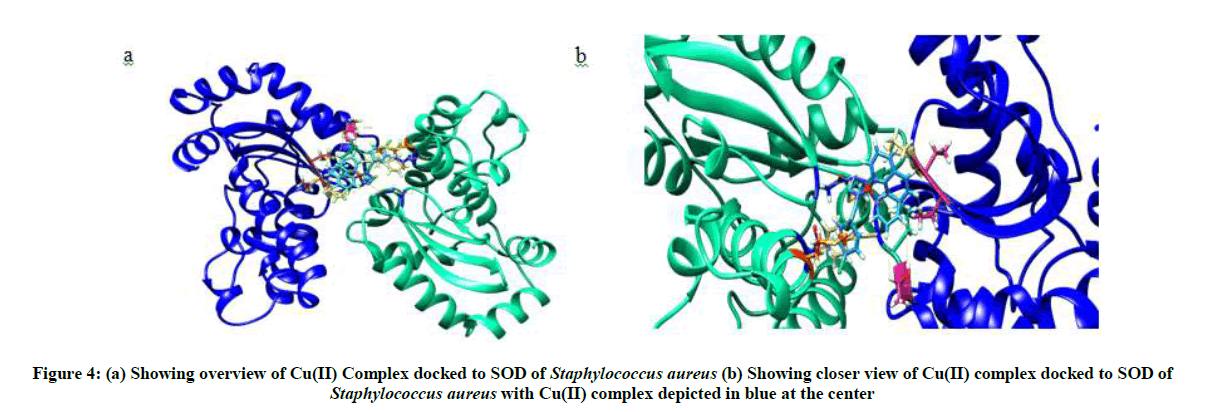
Figure 4: (a) Showing overview of Cu(II) Complex docked to SOD of Staphylococcus aureus (b) Showing closer view of Cu(II) complex docked to SOD of Staphylococcus aureus with Cu(II) complex depicted in blue at the center
Visualization
Visualization of Cu(II) complex docked to SOD in chimera showed 10 interactions with 9 amino acid residues of SOD (Figures 5 and 6).
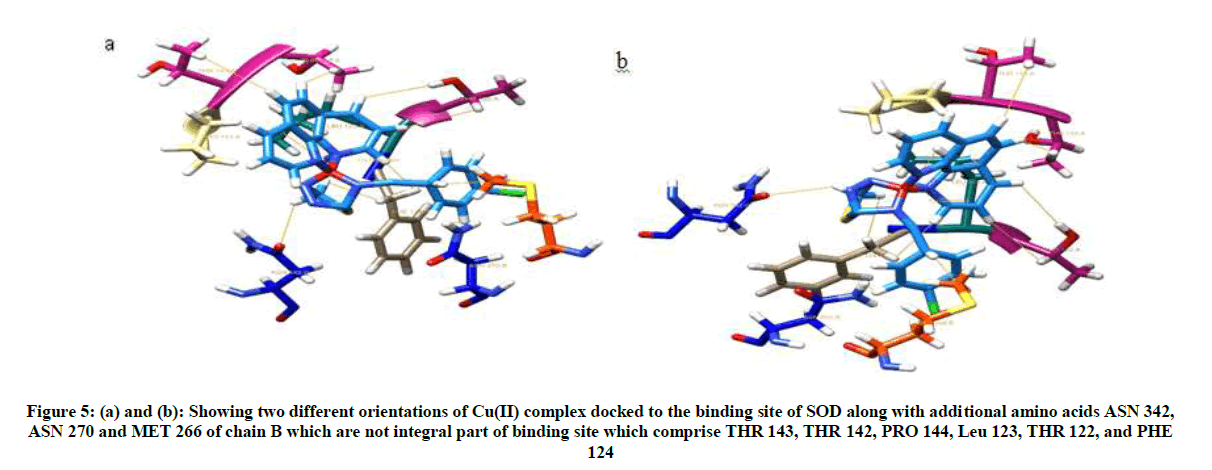
Figure 5: (a) and (b): Showing two different orientations of Cu(II) complex docked to the binding site of SOD along with additional amino acids ASN 342, ASN 270 and MET 266 of chain B which are not integral part of binding site which comprise THR 143, THR 142, PRO 144, Leu 123, THR 122, and PHE 124
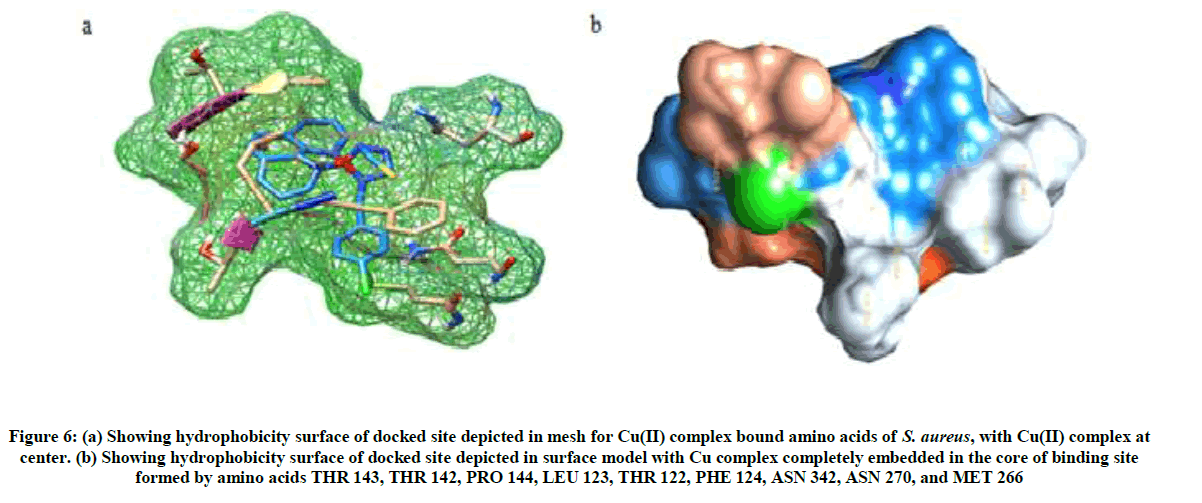
Figure 6: (a) Showing hydrophobicity surface of docked site depicted in mesh for Cu(II) complex bound amino acids of S. aureus, with Cu(II) complex at center. (b) Showing hydrophobicity surface of docked site depicted in surface model with Cu complex completely embedded in the core of binding site formed by amino acids THR 143, THR 142, PRO 144, LEU 123, THR 122, PHE 124, ASN 342, ASN 270, and MET 266
Conclusion
In summary, the Cu(II) complex shows high affinity towards SOD binding site, preventing the binding of its natural substrate. It acts as a non-competitive inhibitor of SOD thereby preventing S. aureus from immune evasion. This paves way for effective treatment of sepsis caused by S. aureus.
Acknowledgement
Authors acknowledge the Head, JSS Academy of Higher Education, Mysuru for computational facilities. C. S. greatly acknowledges the funding support from DST DST-SERB (YSS/2015/001135/LS (Ver-I)).
References
- K.Y. Djoko, M.M. Goytia, P.S. Donnelly, M.A. Schembri, W. M. Shafer, A.G. McEwan, Antimicrob. Agents Chemother., 2015, 59, 6444.
- S.R.M. Mark D. Scott, John W. Eaton, J. Bio. Chem., 1987, 262, 3640.
- J.L.F. Kate, L.R. Dunn, R. Paul Langford, J. Simon Kroll, Infec. Immun., 2003, 71, 1604.
- R.M. Naveen Kumar, K. Shiva Prasad, N. Prasad, Chem. Sci. Trans., 2015, 4, 677.
- B.S. Biasini, A. Waterhouse, K. Arnold, G. Studer, T. Schmidt, F. Kiefer, T.G. Cassarino, M. Bertoni, L. Bordoli, T. Schwede, Nuc. Acids Res., 2014, 252.
- A.K. Kiefer, M. Künzli, L. Bordoli, T. Schwede, Nuc. Acids Res., 2009, 37, 387.
- B.L. Arnold, J. Kopp, T. Schwede, Bioinformatics., 2006, 22, 195.
- P.M.C. Guex, T. Schwede, Electrophoresis., 2009, 162.
- Y.Z. Srayanta Mukherjee, Structure., 2011, 19, 955.
- Z.J. Pintilie, T.D. Goddard, W. Chiu, D.C. Gossard, J. Struct. Biol., 2010, 3, 427.
- A.R. Jianyi Yang, Yang Zhang, Bioinformatics., 2013, 29, 2588.
- A.R. Jianyi Yang, Yang Zhang, Nuc. Acids Res., 2013, 41, 1096.
- S.C. Lovell, W.B. Arendall, J.M. Word, M.G. Prisant, J.S. Richardson, D.C. Richardson, Str. Func. Gen., 2002, 50, 437.
- G.T. Hertig, G.T. Johnson, T.E. Ferrin, Biophys. J., 2015, 9, 2097.
- Combined Quantum Mechanical and Molecular Mechanical methods 1997.
- R.N. Humkey, G.D. Purvis, M.A. Thompson, Nigel Richards, J. Med. Chem., 2005.
- D. Feller, A. Rick Kendall, J. Phys. Chem., 1997, 101, 7292.
- M.A. Thompson, J. Am. Chem. Soc., 1995, 117, 11341.
- M.A. Thompson, Int. J. Quantum Chem., 1996, 60, 1133.
- M.A. Thompson, Proceedings from the 34th Hanford Symposium on the Environment, 1996.
- F.M.J. Brooks.B. R, Gao. J, and Mark A. Thompson Hybrid Quantum and Classical Mechanical Methods for Studying Biopolymers in Solution, in: Pacific Symposium on Biocomputing Eds. L. Hunter and T. E. Klein, World Scientific, New Jersey., 1996.
- H.C. Goddard, T.E. Ferrin, Structure., 2005, 3, 473.
- G.T. Pettersen, C.C. Huang, G.S. Couch, D.M. Greenblatt, E.C. Meng, T.E. Ferrin, J. Comput Chem., 2004, 13, 1605.
- H.C. Chen, T.E. Ferrin, Bioinformatics., 2015, 9, 1484.
- M.E. Huang, J.H. Morris, E.F. Pettersen, T.E. Ferrin, Nuc. Acids Res., 2014, 42, 478.
- L.K. Yang, J. Str. Biol., 2012, 3, 269.
- H.C. Morris, P.C. Babbitt, T.E. Ferrin, Bioinformatics., 2007, 17, 2345.
- H.C. Goddard, T.E. Ferrin, J. Struct. Biol., 2007, 1, 281.
- P.E. Meng, G.S. Couch, C.C. Huang, T.E. Ferrin, BMC Bioinformatics., 2006, 7, 339.
- H.D. Couch, T.E. Ferrin, Nuc. Acids Res., 2006, 34, 29.



