Research - Der Pharma Chemica ( 2023) Volume 15, Issue 3
Microwave-assisted multicomponent synthesis of Pyrrolo[2,3-d] pyrimidinone derivatives and their DFT calculations
Ashish Kumar Panda1, Ruchi Bharti1*, Ajay Thakur1, Monika Verma1 and Renu Sharma1Ruchi Bharti, Department of Chemistry, UIS, Chandigarh University, Mohali, Punjab-140413, India,
Received: 01-May-2023, Manuscript No. dpc-23-99431; Accepted Date: May 29, 2023 ; Editor assigned: 05-May-2023, Pre QC No. dpc-23-99431; Reviewed: 19-May-2023, QC No. dpc-23-99431; Revised: 26-May-2023, Manuscript No. dpc-23-99431; Published: 31-May-2023, DOI: 10.4172/0975-413X.15.3.52-61
Abstract
Herein, we have reported a green and environmentally benign methodology for synthesizing novel pyrrolo[2,3-d] pyrimidine derivatives under microwave-assisted reaction conditions. The desired product was obtained with an excellent yield (80-90%) within 30 minutes only. The characteristic features of this protocol were the use of green reaction conditions, high atom economy, less reaction time, excellent yields, no use of specific purification processes, no column chromatography, etc. Later, theoretical studies like DFT were also performed on each newly synthesized product (4a-e) using B3LYP (Becke-Lee-Yang-Parr) functional and 6-31+G (d,p) to get spot onto the geometry, electronic transitions, and spectroscopic properties theoretically that has been compared with the encountered experimental ones. The molecular electrostatic potential surface, Band gap (δE), Chemical Potential (μ), Chemical hardness (n), Ionization Potential (I), Electron Affinity (A), Electronegativity (X), potential, Electrophilicity index (ω) are computed in order to find out electrophilic and nucleophilic sites. All the newly synthesized pyrrolo[2,3-d] pyrimidine derivatives were subjected to DPPH, ABTS, and TAC for antioxidant activities. 4b and 4d showed maximum potent antioxidant activity
Keywords
Pyrrolo pyrimidinone; Microwave irradiation; Multicomponent reactions; DFT analysis; Anti-oxidant activity
INTRODUCTION
Pyrimidinone is a nitrogen-containing heterocyclic compound structure shown in (Figure 1) that has acquired an important position due to its different physiochemical properties in the medicinal industry [1]. It acts as one of the critical elements in RNA and DNA nucleobases. In addition, pyrimidinones have many biologically active molecules, vital in clinical candidates [2].

Figure 1. Pyrimidinone
They have diverse biological activities such as analgesic [3], anti-microbial [4], anti-inflammatory [5], anti-viral [6], antioxidant [7], anti-fungal [8], anti-malarial [9] and anti-angiogenetic agents [10]. Here are a few examples of pharmaceutical active pyrimidinone compounds (Figure 2).

Figure 2. Biological active pyrrolo [2, 3-d] pyrimidine derivatives
Due to their significant biological activities, fused pyrimidinone has attracted the interest of many researchers for the past few years [11-13].
Further, based on efficiency, convergence, simplicity, and selectivity, recently, multicomponent reactions have been the most widely used platform for synthesizing complex organic compounds [14]. These reactions are vital in medicinal chemistry, the pharmacy industry, and drug development [15]. Also, in the past few years, microwave-assisted reactions have gained much popularity in organic synthetic reactions because of taking less time to complete the reaction and reducing the energy consumption required for heating [16].
Considering all these facts, and also because of their proven biological activities and medicinal utilities, many efforts have been made toward the molecular manipulation of this molecule. However, from the literature survey, it has been observed that only a few methods have been reported for the synthesis of pyrrolo [2, 3-d] pyrimidines [17] having 1,3 diketones and 6-amino uracil moieties. The above synthesis reaction was reported by Secrist et al. [18], which was further modified by Kretschmer [19]. However, no direct report on synthesizing pyrrolo [2, 3-d] pyrimidines with 1, 3-diketones and 5-amino uracil has yet been reported.
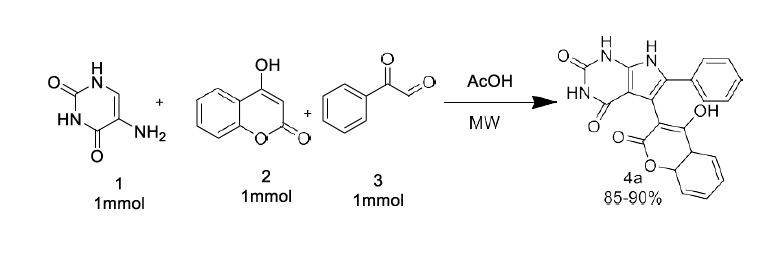
As a part of our continued interest in synthesizing different heterocyclic compounds of biological importance [20-23], we developed an effective method for the synthesis of pyrrolo[2,3-d] pyrimidinone derivatives by the reaction of N, N-dimethyl 5 amino uracil (1), 1,3-diketone (2) and Phenyl glyoxal monohydrate derivatives (3) under microwave irradiation using acetic acid as a solvent system.

Scheme 1: General Reaction
Initially, 1 mmol of 5 amino uracils (1) was reacted with 4-hydroxy coumarin (2), and phenyl glyoxal monohydrate (3) was taken as a model reaction under solvent acetic acid and catalyst-free conditions. The effect of solvent, catalyst, and reaction condition was changed carefully for the given model reaction. Earlier, when the reaction was performed at room temperature in the reaction condition, we could not isolate any product even after 24 hours (Table 1) (Entry 1).
Later the condition was changed, and it was observed that under reflux conditions, acetic acid: H2O in (1:1) ratio gave a higher yield of 40% within 10 hours. (Table 1) (Entry 2). Different solvents like ethanol and water, AcOH were also used under different reaction conditions, which provided higher yields of 65-74% of our desired products (Table 1) (Entries 4-7). Further, we carried out the exact model reaction in MW using a solvent system of acetic acid and ethanol in an equimolar ratio of 1:1. (Entry 8). To our surprise, the targeted product, 4a, was observed with an excellent yield of 92% yields within half an hour. Later, we explored other solvent systems but ended with a lower yield only (Entries 9-10). The formation of pyranopyrazole derivative 4a was confirmed by IR, 1H-NMR, and 13C-NMR analysis, which show good agreement with all the spectra.
| SN. | Solvent | Conditions | Time (hrs.) | Yieldc(percentage) |
|---|---|---|---|---|
| system | ||||
| 1 | AcOH | Room | 24 | Traces |
| Temperature | ||||
| 2 | AcOH | Reflux | 10 | 40 |
| condition | ||||
| 3 | Ethanol | Reflux | 10 | 54 |
| condition | ||||
| 4 | H2O | Reflux | 10 | 65 |
| condition | ||||
| 5 | AcOH | Microwave | 0.5 | 74% |
| 6 | H2O | Microwave | 0.5 | 72 |
| 7 | Ethanol | Microwave | 0.5 | 70 |
| 8 | AcOH: H2O |
Microwave | 0.5 | 92 |
| (1:1) | ||||
| 9 | AcOH: H2O |
Ultrasonic | 1 | 78 |
| (1:1) | sonicator | |||
| 10 | AcOH: Ethanol |
Microwave | 0.5 | 68 |
| (1:1) |
Under optimized conditions, we further explored the effectiveness of this method by changing the substrates of the reactants. For this different 1,3- diketones as hydroxynaphthaquinone and 4-hydroxycoumarin were treated with various substituted ayl glyoxal and 5-aminouracil to afford the pyranopyrazole derivatives under microwave irradiation conditions. All the reactions went smoothly and provided very good to exceptional yields of the required products without significantly affecting the total yield and reaction time (Table 2).

Table 2: Scope of the reactiona
On the basis of experimental results, a reasonable mechanism has been proposed for the synthesis of compound 4a in scheme 3. At first, a Knoevenagel condensation of 4-hydroxy coumarin 2 with Phenyl glyoxal monohydrate 3 occurs to give an intermediate A. Further 5- amino uracil 1 undergoes nucleophilic addition with the intermediate A to provide the intermediate B. Then the intermediate undergoes intramolecular cyclization by losing a H2O molecule to produce the desired compound 4a.

Scheme 3: Mechanistic approach for the synthesis of pyrrolo[2,3-d] pyrimidinone derivatives.
DFT Calculations (2a)
DFT calculations were performed using the Gaussian software with the B3LYP/6-311G basis set [24] to investigate the frontier molecular orbitals (FMOs) and ideal structures of compounds 4a-4e. Figure 4 presents the optimized structures along with the corresponding FMOs. Analysis of the FMOs, including the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), provides valuable insights into the chemical reactivity of the compounds. The HOMO serves as an electron donor, whereas the LUMO acts as an electron acceptor. Utilizing the HOMO and LUMO, we further determined the electron affinity, electronegativity, ionization potential, energy band gap, and chemical hardness of the compounds, as summarized in (Table 3) using the equation derived from Koopmans theorem. The equations are as follows:
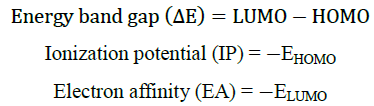

Using HOMO and LUMO, we also calculate the electron affinity, electron negativity, ionization potential, Energy band gap, and chemical hardness, shown in (Table 3)
The energy band gap is the difference between the LUMO and HOMO of the molecule, which gives information about the reactivity of the molecule Figure 4(a-e)
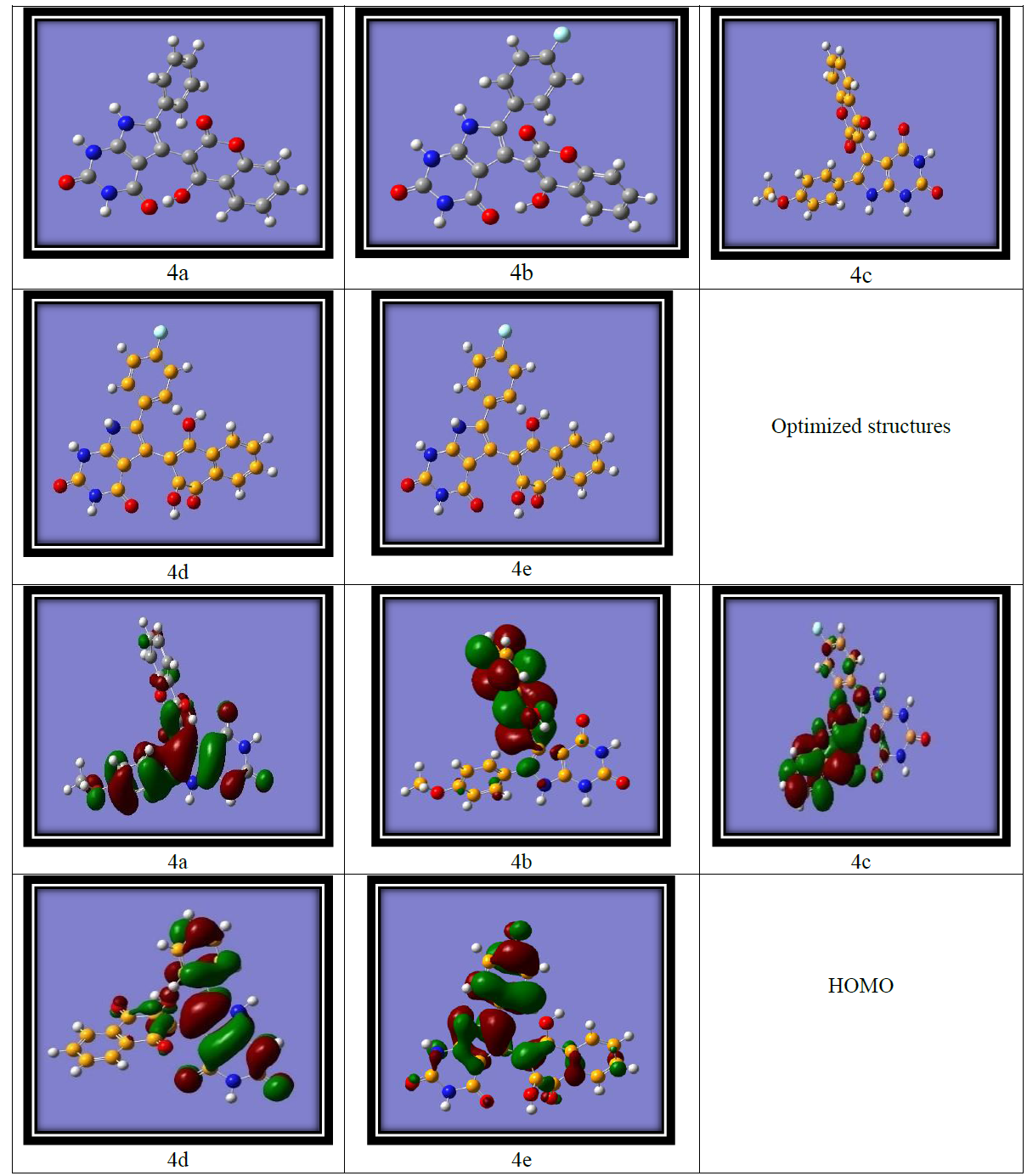

Figure 4(a-e): HOMO LUMO of Synthesized molecules
The energy band gap, defined as the difference between the LUMO and HOMO energies, provides insights into the reactivity of the molecules. (Table 3) (entry 3) displays the energy gaps between the molecules. Among the synthesized compounds, compound 4a exhibits the lowest band gap, measuring 4.08 eV, while compound 4e demonstrates the highest energy gap at 8.11 eV. This suggests that charge transfer and electron transition activities occur more readily in compound 4a, where the band gap is minimal. Conversely, compound 4e, with the largest band gap, experiences greater difficulty in charge transfer activities. Further, electron affinity, highest for compound 4b (3.34) was observed, indicates that it has a greater tendency to accept electrons compared to the other compounds. The electron affinity refers to the energy change that occurs when an electron is added to a neutral atom or molecule to form a negative ion. It represents the tendency of a species to accept an electron. A higher electron affinity value indicates a greater ability to accept electrons. The Mullikan electron negative value reveals that compound 4e possesses the highest electronegativity (5.15), indicating a greater propensity to accept electrons. Furthermore, the quantum mechanical descriptor known as the chemical potential can be used to predict the molecular stability. Notably, compound 4e exhibits the highest negative value in the table, suggesting its high stability under normal conditions.
| Sr. No. | Quantum mechanical descriptors | 4a | 4b | 4c | 4d | 4e |
|---|---|---|---|---|---|---|
| 1. | HOMO Energy (eV) | -5.96 | -5.88 | -8.2 | -6.49 | -9.21 |
| 2. | LUMO Energy (eV) | -1.88 | -3.34 | -1.84 | -0.76 | -1.1 |
| 3. | Band gap (eV) | 4.08 | 5.54 | 6.36 | 5.73 | 8.11 |
| 4. | Chemical potential (μ) | -3.92 | -3.11 | -5.02 | -3.48 | -5.15 |
| 5. | Chemical hardness (η) | 2.04 | 2.77 | 3.18 | 2.86 | 4.05 |
| 6. | Ionization potential (IP) | 5.96 | 5.88 | 8.2 | 6.49 | 9.21 |
| 7. | Electron affinity (EA) | 1.88 | 3.34 | 1.84 | 0.76 | 1.1 |
| 8. | Electron negativity (X) | 3.92 | 3.11 | 5.02 | 3.48 | 5.15 |
| 9. | Electrophilicity index (ω) | 3.76 | 1.74 | 3.96 | 2.11 | 3.27 |
Antioxidant Activity (2b)
The DPPH, TAC, and ABTS methods were used to determine the antioxidant activity of the pyrimidinone derivatives (4a-4e) that we prepared. The outcomes of the antioxidant activity are shown in (Table 4)
| Sample | DPPH | ABTS | TAC |
|---|---|---|---|
| PERCENTAGE (%) | |||
| 4a | 5.02 | 49.39 | 13.69 |
| 4b | 9.07 | 43.97 | 10 |
| 4c | 8.68 | 49.48 | 43.99 |
| 4d | 11.48 | 37.45 | 17.58 |
| 4e | 17.75 | 29.9 | 11.59 |
| Ascorbic acid | 95 | 95 | 92 |
| Gallic acid | 82.5 | 82.5 | 84 |
Any organic compounds antioxidant activity mainly depends upon the ability to donate its electron to DPPH radicals. When a molecule donates its electron to the DPPH radicals, it converts into a diamagnetic frame. When the DPPH solution changes colour, it indicates the interaction between pyrimidinone derivatives and DPPH free radicals. When the dark violet colour changes to brown colours, it indicates the pyrimidinone derivatives' oxidation potential. Results of the scavenging potential of the compounds revealed that these compounds are showing inhibition. Compound 4e has maximum inhibition, i.e., 17.75%, but it had a lower inhibition than the standards. All the compounds show antioxidant activity in this activity, but it was very low.
ABTS free radicals scavenge any substance with antioxidant capability. When we scavenged ABTS free radicals, we found a decrease in the sample's absorption. The sample activity was done at 734 nm, and the results were compared with the standard solutions and analyzed. When the ABTS free radicals start interacting with the synthesized molecules, then the color of the solution starts changing. After the evaluation of the results, it is observed that compound 4c shows the highest antioxidant potential during this process. The rest of the compounds 4a, 4b, 4d, and 4e show moderate antioxidant potential.
Antioxidants will cause the phosphomolybdate ions to be reduced and create a green molybdate complex, which was measured at 695nm using a Spectrophotometer. In this method, the derivative 4c, with a value of 43.99%, demonstrated the highest overall antioxidant capacity.
Conclusion
In conclusion, we have developed a clean, simple, one-pot three-component reaction for synthesizing pyrrolo[2,3-d] pyrimidine-2,4(3H)-Dione derivatives in AcOH: H2O. 1H NMR, FT-IR, and 13C-NMR were used to identify the structure of the synthesized compounds. This reaction involves a few characteristic features, such as using a non-conventional energy source, simple reaction conditions, very little time, and a general reaction procedure. The synthesized compounds were evaluated for their antioxidant assay and showed positive results. Also, density functional calculations are used to evaluate the HOMO-LUMO energy gap and the molecular electrostatic potentials, electronegativity and ionization potential.
EXPERIMENTAL SECTION
Tetramethyl silane was used as the internal standard for the NMR study, which was performed using a Bruker Avance II 500 MHz NMR spectrometer in CDCl3 and DMSO-d6. The IR spectra of each compound were measured with the help of the Shimadzu FTIR spectrophotometer. Melting points of synthesized analogs were calculated on digital melting point equipment and are uncorrected. Chemical shift values were assessed in TLC aluminum plates covered with silica gel in δ (ppm). A Perkin Elmer 2400 automated carbon, hydrogen, and nitrogen analyzer was used to conduct the elemental analyses. The melting points and data from the literature were compared to all known compound data.
General procedure for the preparation of 4a-e (3a)
At first, the reaction mixture of equimolar of 5-amino uracil, 4-hydroxycoumarin, and aryl glyoxal was irradiated in AcOH: H2O medium (1:1) for 30 minutes. Later, TLC was used to track the reaction's progress (Hexane: Ethyl Acetate; 70:30). Finally, the water was subsequently used to wash the reaction mixture. Finally, the solid product obtained through vacuum-filtered was recrystallized with hot ethanol to produce the pure product.
Characterization and Spectral Analysis of Product 4a (3b)
4a: White solid, Melting point (>2500C) 1H NMR (500MHz, DMSO-d6): δ = 12.28 (1H, S, OH), 11.27 (1H, S, NH), 10.76 (1H, S, NH), 10.67 (1H, S, NH), 7.88 (1H, d, J=10Hz, ArH), 7.65(1H, t, J=10Hz), 7.42(1H, d, J=10Hz, ArH), 7.38(S, 1H, ArH), 7.34(t, 2H, J=10Hz, ArH), 7.29-7.26(m, 1H, ArH) ppm; 13CNMR (500MHz, DMSO-d6): δ= 162.90, 162.10, 155.79, 152.81, 151.59, 138.58, 136.15, 132.23, 131.37, 128.48, 128.04, 126.78, 123.73, 123.66, 116.40, 116.11, 110.52, 97.62, 96.92, 39.75, 39.58, 39.25, 39.08 ppm; Elemental analysis calculated(M.W; 389.37) C,64.78; H, 3.88; N, 10.79; O,20.54; Elemental analysis (found) C, 64.83; H, 3.85; N, 10.90; O, 20.54.
ACKNOWLEDGMENTS
The authors are grateful to the Department of Chemistry, Chandigarh University, for providing the basic facilities and resources. For providing the analytical facilities to characterize compounds, the authors thank SAIF- Punjab University.
REFERENCES
- Baraldi PG, Tabrizi MA, Gessi S, et al. Chem. Rev. 2008, 108:p.238.
- Petricci E, Mugnaini C, Radi M, et al. Arkivoc. 2006, 7:p.452.
- Manlove A, Groziak MP. Eds. 2009, 20: p. 333.
- Chamberlain SD, Wilson JW, Deanda F et al. Bioorg. Med. Chem. Lett. 2009, 19: p. 469.
- Tangeda SJ, Garlapati A. Eur. J. Med. Chem. 2010, 45:p. 1453.
- Mohamed MS, Kamel R, Fatahala SS. Eur. J. Med. Chem. 2010, 45:p. 2994.
- Hilmy KMH, Khalifa MMA, Hawata MA et al. Eur. J. Med. Chem. 2010, 45:p. 5243.
- Ghorab MM, Ragab FA, Heiba HI et al. Bioorg. Med. Chem. Lett. 2010, 20:p. 6316.
- McHardy T, Caldwell JJ, Cheung KM et al. J. Med. Chem. 2010, 53: p. 2239.
- Gangjee A, Yang JJ, McGuire RL et al. Bioorg. Med. Chem. 2006, 14: p. 8590.
- Janeba Z, Balzarini J, Andrei G et al. J. Med. Chem. 2005, 48: p. 4690.
- Gangjee A, Kurup S, Ihnat MA et al. Bioorg. Med. Chem. 2010, 18: p. 3575.
- Varaprasad CVNS, Ramasamy SK, Girardet JL et al. Bioorg. Chem. 2007, 35: p. 25.
- Koh YH, Shim JH, Girardet JL et al. Bioorg. Med. Chem. Lett. 2007, 17: p. 5261.
- Moriarty KJ, Koblish HK, Garrabrant T et al. Bioorg. Med. Chem. Lett. 2006, 16: p. 5778.
- Shaker RM, Abd Elrady MZ, Naturforsch. 2008, 63b: p. 1431.
- Shaker RM, Sadek KU, Hafez EA et al. 2010, 65b:p. 1485
- Shaker RM, Ameen MA, Abdel Hameed AM et al. Tetrahedron Lett. 2010, 52:p. 6319.
- Shaker RM, Ibrahim YR, Abdel-Latif FF et al. Naturforsch. 2010, 65b:p. 1148.
- Thakur, Ajay, Monika Verma et al.Tetrahedron.2022, p. 132813.
- Thakur, Ajay, Ruchi Bharti et al. Orbital: The Electronic Journal of Chemistry. 2021, p.335-349.
- Kumari, Pooja, Ruchi Bharti. Molecular Diversity. 2019, 23:p. 205-213.
- Verma, Monika, Ajay Thakur et al. Current Organic Synthesis. 2022, 19:p. 86-114.
- Kuruvilla, Tintu K. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy.2018,188:p. 382-393.
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref



