Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 10
Investigation of Spectroscopic, Electronic and Geometric Properties of 2-(3-Methyl-4,5-Dihydro-1h-1,2,4-Triazol-5-On-4-Yl)-Azomethine)-Benzoic Acid using B3lyp and Hf Basis Sets
Gül Kotan1*, Gül Özdemir2 and Haydar Yüksek2
1Kafkas University, Kars Vocational School, Kars, Turkey
2Kafkas University, Department of Chemistry, Kars, Turkey
Abstract
2-(3-Methyl-4,5-dihydro-1H-1,2,4-triazol-5-on-4-yl)-azomethine)-benzoic acid has been optimized using B3LYP/6-31G(d) and HF/6-31G(d) basis sets. Thus, the most stable geometrical comformer of compound was obtained. Proton Nuclear Magnetic Resonance (1H-NMR) and Carbon-13 Nuclear magnetic Resonance (13C-NMR) spectral values according to GIAO method was calculated using Gaussian G09W program package in gas phase and in DMSO solvent. Theoretical and experimental values were plotted according to δ exp=a+b.d calc. Theoretical spectral values of molecule were calculated and compared with experimental values. The veda4f program was used in defining Infrared (IR) data. The standard error values were found via the Sigma plot with regression coefficient of a and b constants. The vibrational frequency values of this molecule have been calculated by using 6-31G (d) basis set with DFT and HF methods. Then, these values are multiplied with appropriate adjustment factors. In addition, the, thermodynamics properties (heat capacity CV0, entropy S0 and enthalpy H0), electronic properties (electronegativity (χ), electron affinity (A), global hardness (η), softness (σ), ELUMO-EHOMO energy gap (ΔEg) and ionization potential (I), HOMO-LUMO energy), geometric properties (bond angles, bond lengths), dipole moments, mulliken atomic charges, total energy of the molecule were calculated with Gaussian 09W program on the computer.
Keywords
B3LYP, HF, GIAO, Mulliken charge, Veda4f.
Introduction
Schiff bases have an azomethine group (-CH=N-) which is synthesis by the condensation of a primary amine with a carbonyl compound. Schiff base compounds indicate a sort of biological properties [1,2] and have been used as bacteriocides, insecticides, pesticides, and fungicides [3,4]. Besides, the compounds including 1H-1,2,4-triazol group and their derivatives have very imported biological activities such as antioxidant [5-7] antibacterial [8-12], antifungal [13], anti-inflammatory [14], anti-HIV [15], antitumor [16], anticonvulsant [17], pharmacological activity [18], antiviral [12], antihypertensive [19], antitubercular [20] and analgesic [21,22] properties. In this study, we then investigated theoretically the structural, molecular, electronic, thermodynamics and vibrational properties of the synthesized compound. Experimental data obtained from the literature [23]. All quantum chemical calculations were conducted with the GaussView and Gaussian 09 packet programs [24,25]. For this, firstly, 2-(3-Methyl-4,5-dihydro-1H-1,2,4-triazol-5-on-4-yl)-azomethine)-benzoic acid molecule was optimized by using 631G (d,p) basis set of Density Functional Theory (DFT/B3LYP) and Hatree Fock (HF) methods. The subsequent theoretical calculations were made by targeting this optimized geometry which from this the most stable structure of the molecule. The optimized structure along with the atom numbering is given in Figure 1. Furthermore, the 1H and 13C-NMR chemical shifts (Table 1) calculations were performed using the Gauge-Independent Atomic Orbital (GIAO) [26] with B3LYP/631G(d,p) and HF/631G(d,p) levels in ethanol. Theoretical and experimental values were inserted into the grafic according to equatation of δ exp=a+b. δ calc. The standard error values were found via Sigma Plot program with regression coefficient of a and b constants. The veda4f program [27] was used in defining of IR data theoretically and the theoretical vibrational (Table 2) spectra were calculated at the B3LYP/6-31G (d) in the gas phase, then scaled by 0.9617 and 0.8992 [28], while an identical scaling factors of 0.9617 and 0.8992 were applied for all B3LYP/DFT/HF6-31G (d) frequencies [29]. The data obtained according to calculate thereotically are formed using theoretical infrared spectrum. The experimental and theoretical IR spectra are given in Figure 2. The experimental and the calculated theoretical spectral values were compared and they were recorded to be compatible with each other. In addition to, geometric properties (bond angles, bond lengths and dihedral angles) (Tables 3 and 4), electronic properties (ELUMO-EHOMO) energy gap (ΔEg), electronegativity (χ), electron affinity (A), global hardness (η), softness (σ), ionization potential (I), total energy of the molecule, dipole moment) (Figures 3-7 and Tables 5-8), thermodynamics properties, the highest occupied molecular orbital (HOMO) (Figure 8) and the lowest unoccupied molecular orbital (LUMO) (Figure 8), mulliken atomic charges have been examined by using Gaussian 09W program.
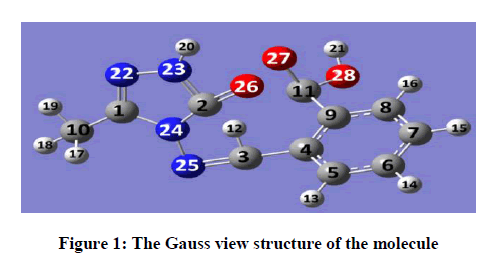
Figure 1: The Gauss view structure of the molecule
| No. | Exp. | DFT/dmso | Differ/DFT | HF/dmso | Differ/HF |
|---|---|---|---|---|---|
| C1 | 144.34 | 150.39 | -6.05 | 145.27 | -0.93 |
| C2 | 151.02 | 152.01 | -0.99 | 145.61 | 5.41 |
| C3 | 152.48 | 156.71 | -4.23 | 153.80 | -1.32 |
| C4 | 133.61 | 142.23 | -8.62 | 135.24 | -1.63 |
| C5 | 126.79 | 132.07 | -5.28 | 125.35 | 1.44 |
| C6 | 132.01 | 137.56 | -5.55 | 132.65 | -0.64 |
| C7 | 130.71 | 134.78 | -4.07 | 126.25 | 4.46 |
| C8 | 131.52 | 137.02 | -5.50 | 130.80 | 0.72 |
| C9 | 130.17 | 132.37 | -2.20 | 123.14 | 7.03 |
| C10 | 11.06 | 22.90 | -11.84 | 11.80 | -0.74 |
| C11 | 167.75 | 168.64 | -0.89 | 158.67 | 9.08 |
| H12 | 10.39 | 10.83 | -0.44 | 9.96 | 0.43 |
| H13 | 7.91 | 8.56 | -0.65 | 8.00 | -0.09 |
| H14 | 7.65 | 8.13 | -0.48 | 7.97 | -0.32 |
| H15 | 7.62 | 8.00 | -0.38 | 7.76 | -0.14 |
| H16 | 8.01 | 8.63 | -0.62 | 8.45 | -0.44 |
| H17 | 2.26 | 2.71 | -0.45 | 2.00 | 0.26 |
| H18 | 2.26 | 2.86 | -0.60 | 2.22 | 0.04 |
| H19 | 2.26 | 2.42 | -0.16 | 1.83 | 0.43 |
| H20 | 11.82 | 7.41 | 4.41 | 6.56 | 5.26 |
| H21 | 11.82 | 6.67 | 5.15 | 5.83 | 5.99 |
Table 1: The calculated and experimental 13C and 1H-NMR DMSO (B3LYP/HF 631G (d) isotropic chemical shifts
| Vibration | Experimental | Theoretical DFT/HF |
|---|---|---|
| ν (OH) | 3528 | 3553/3622 |
| ν (NH) | 3302 | 3541/3520 |
| ν (C=N) | 1614 | 1610/1696 |
| ν (C=O) | 1695-1652 | 1741/1792 |
Table 2: Significant vibrational frequencies (cm-1) of the molecule
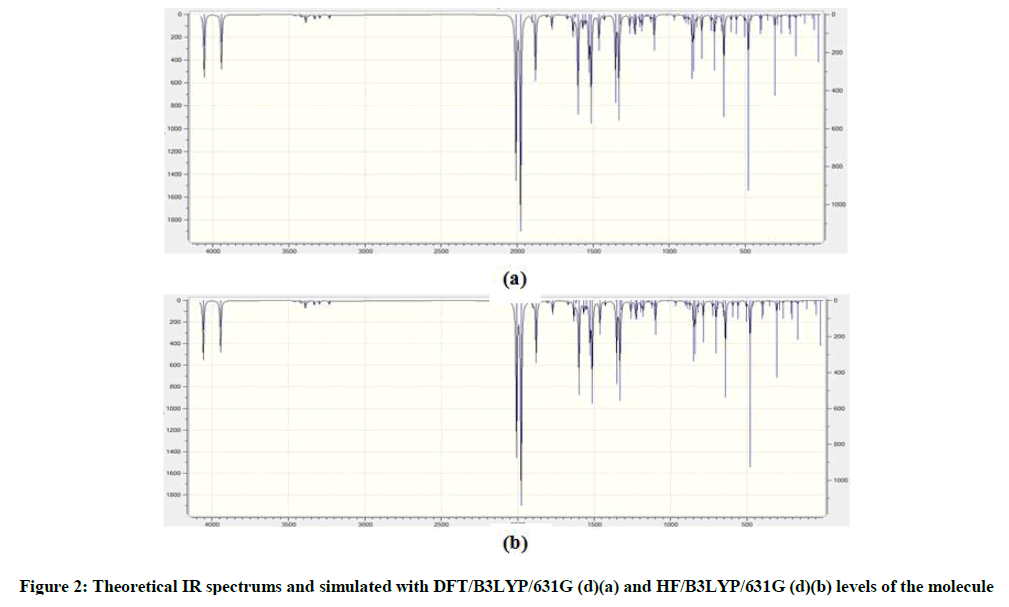
Figure 2: Theoretical IR spectrums and simulated with DFT/B3LYP/631G (d)(a) and HF/B3LYP/631G (d)(b) levels of the molecule
| Bağ Açıları | B3LYP | HF | Bağ Açıları | B3LYP | HF |
|---|---|---|---|---|---|
| N(22)-C(1)-N(24) | 111.44 | 111.32 | C(3)-C(4)-C(5) | 121.42 | 118.63 |
| N(22)-N(23)-H(20) | 120.24 | 120.75 | C(3)-C(4)-C(9) | 122.57 | 122.68 |
| C(22)-N(23)-C(2) | 114.64 | 113.81 | C(4)-C(5)-H(13) | 118.04 | 118.77 |
| N(23)-C(2)-O(26) | 130.11 | 129.63 | C(4)-C(5)-C(6) | 121.42 | 121.03 |
| O(26)-C(2)-N(24) | 128.91 | 128.61 | C(5)-C(6)-H(14) | 119.74 | 119.63 |
| N(23)-C(2)-N(24) | 100.97 | 101.75 | C(5)-C(6)-C(7) | 120.05 | 120.18 |
| N(22)-C(1)-C(10) | 125.12 | 125.46 | H(14)-C(6)-C(7) | 120.20 | 120.17 |
| C(1)-C(10)-H(17) | 111.09 | 110.58 | C(6)-C(7)-H(15) | 120.47 | 120.48 |
| C(1)-C(10)-H(18) | 111.02 | 110.57 | H(15)-C(7)-C(8) | 119.96 | 119.97 |
| C(1)-C(10)-H(19) | 108.67 | 108.66 | C(7)-C(8)-H(16) | 120.16 | 119.95 |
| C(1)-N(24)-C(2) | 108.41 | 108.32 | C(7)-C(8)-C(9) | 120.97 | 120.72 |
| C(24)-N(25)-C(3) | 118.41 | 118.64 | C(9)-C(11)-O(27) | 126.08 | 125.13 |
| N(25)-C(3)-H(12) | 122.60 | 122.87 | C(9)-C(11)-O(28) | 112.63 | 113.25 |
| H(12)-C(3)-C(4) | 119.44 | 119.08 | C(11)-O(28)-H(21) | 105.31 | 107.66 |
Table 3: The calculated bond angles of the molecule
| bond lengths | DFT | HF | bond lengths | DFT | HF |
|---|---|---|---|---|---|
| C(1)-N(22) | 1.300 | 1.269 | C(3)-C(4) | 1.475 | 1.486 |
| C(1)-N(24) | 1.388 | 1.377 | C(4)-C(5) | 1.405 | 1.389 |
| C(1)-C(10) | 1.487 | 1.488 | C(4)-H(13) | 1.084 | 1.072 |
| N(22)-N(23) | 1.381 | 1.371 | C(5)-C(6) | 1.390 | 1.383 |
| N(23)-H(20) | 1.007 | 0.992 | C(6)-H(14) | 1.086 | 1.075 |
| N(23)-C(2) | 1.371 | 1.347 | C(6)-C(7) | 1.396 | 1.383 |
| C(2)-N(24) | 1.421 | 1.387 | C(7)-H(15) | 1.086 | 1.074 |
| C(2)-O(26) | 1.220 | 1.200 | C(7)-C(8) | 1.391 | 1.382 |
| N(24)-N(25) | 1.371 | 1.368 | C(8)-H(16) | 1.083 | 1.071 |
| C(10)-H(17) | 1.095 | 1.083 | C(8)-C(9) | 1.403 | 1.390 |
| C(10)-H(18) | 1.095 | 1.083 | C(8)-C(11) | 1.489 | 1.490 |
| C(10)-H(19) | 1.091 | 1.080 | C(11)-O(27) | 1.215 | 1.190 |
| N(25)-C(3) | 1.290 | 1.259 | C(11)-O(28) | 1.359 | 1.329 |
| C(3)-H(12) | 1.083 | 1.069 | O(28)-H(21) | 0.975 | 0.952 |
Table 4: The calculated bond lengths of the molecule
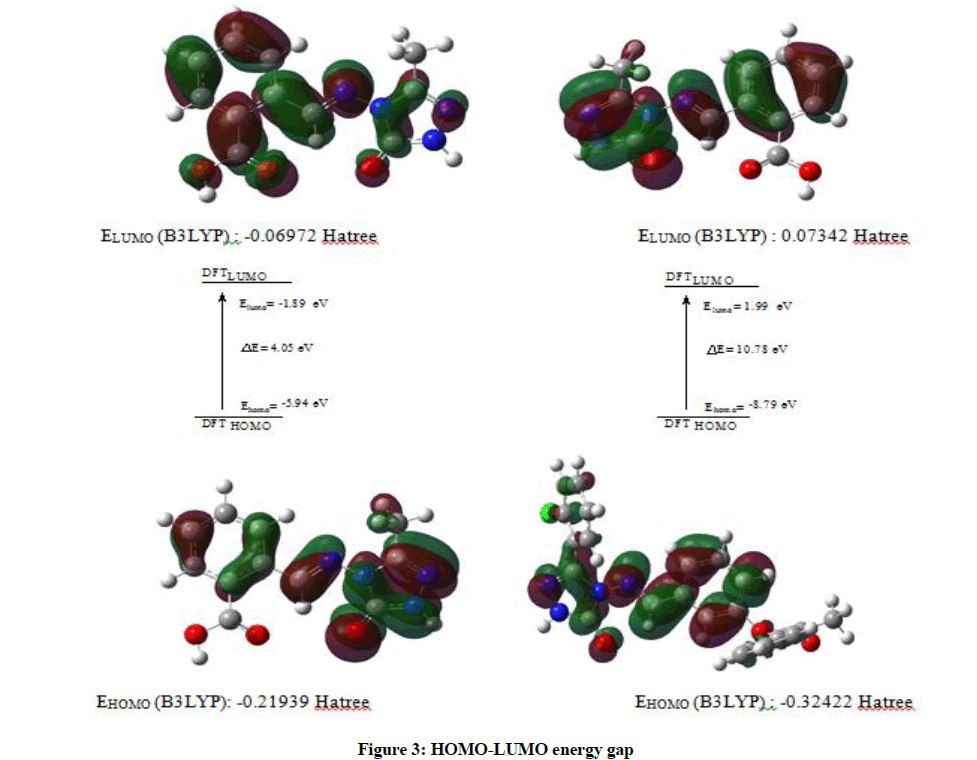
Figure 3: HOMO-LUMO energy gap
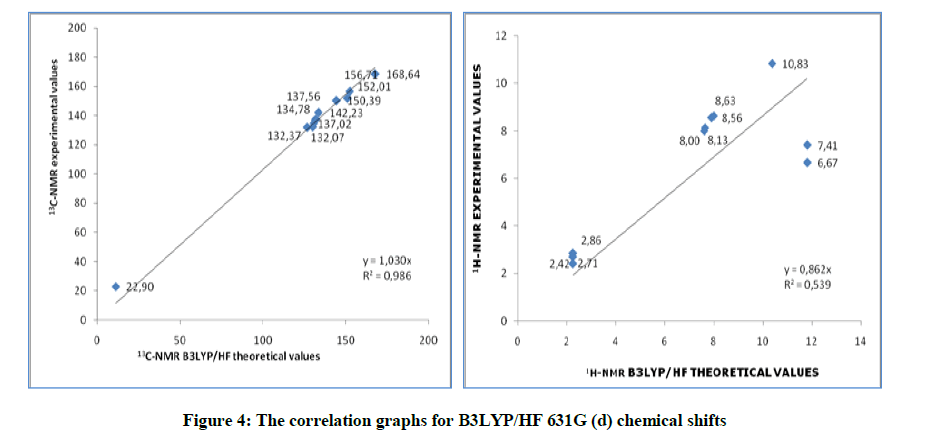
Figure 4: The correlation graphs for B3LYP/HF 631G (d) chemical shifts

Figure 5: The MEP of the molecule
Figure 6: The Total density of the molecule
Figure 7: The ESP of the molecule
| Enerji (a.u.) | DFT | HF |
| -869.8542 | -864.7481 |
Table 5: The calculated total energy datas B3LYP/HF of the molecule
| μx | μy | μz | μToplam | |
|---|---|---|---|---|
| Dft | -1.5832 | 2.8439 | 1.1798 | 3.4622 |
| Hf | 2.4571 | -1.6573 | -2.496 | 3.8748 |
Table 6: The calculated dipole moments datas of the molecule
| DFT | HF | DFT | HF | ||
|---|---|---|---|---|---|
| C1 | 0.544 | 0.607 | H15 | 0.143 | 0.217 |
| C2 | 0.189 | 1.048 | H16 | 0.168 | 0.251 |
| C3 | 0.046 | 0.117 | H17 | 0.183 | 0.203 |
| C4 | 0.078 | -0.018 | H18 | 0.180 | 0.201 |
| C5 | -0.166 | -0.200 | H19 | 0.184 | 0.210 |
| C6 | -0.120 | -0.183 | H20 | 0.356 | 0.417 |
| C7 | -0.126 | -0.204 | H21 | 0.415 | 0.474 |
| C8 | -0.170 | -0.182 | N22 | -0.321 | -0.340 |
| C9 | 0.032 | -0.144 | N23 | -0.519 | -0.659 |
| C10 | -0.507 | -0.522 | N24 | -0.411 | -0.622 |
| C11 | 0.534 | 0.790 | N25 | -0.303 | -0.290 |
| H12 | 0.237 | 0.299 | O26 | -0.531 | -0.651 |
| H13 | 0.159 | 0.238 | O27 | -0.469 | -0.568 |
| H14 | 0.143 | 0.218 | O28 | -0.579 | -0.708 |
Table 7: The calculated mulliken charges datas B3LYP/HF 631G (d,p) of the molecule
| 13C | 1H | |||||||
|---|---|---|---|---|---|---|---|---|
| R | S. hata | a | b | R | S. hata | a | b | |
| DFT | 0.997 | 2.215 | 1.063 | -13.5 | 0.651 | 2.358 | 0.337 | 1.036 |
| HF | 0.992 | 3.837 | 1.024 | -1.047 | 0.598 | 2.53 | 1.303 | 0.973 |
Table 8: The correlation data for chemical shifts
Figure 8: The Electrostatic Potential
Materials and Methods
Method
Computational details
The IR data of the compound
Theoretically IR values were calculation veda4f programme and scala values were obtain. Theoretically calculated IR data are multiplied with appropriate adjustment factors respectively 0.9613, 0.8929 for DFT/631G(d), HF/631G(d) basis sets. All values are found positive. IR spectrums were drawn with obtained values according to HF and DFT method (Figure 2). Experimentally IR values were compared with theoretically IR values. In this compared were estimated corresponding with each other of values.
The relation between R values
B3LYP/631G (d,p) (DMSO): 13C: 0.997, 1H: 0.651; HF/631G (d,p) (DMSO): 13C: 0.992, 1H: 0.598. There is such a relationship between R-values of the compound. Found standard error rate R and a, b constants regression values were calculated according to formule exp =a + b. δ calc Eq. These values for compound were summarized in the Table 9. Theoretical and experimental 1H and 13C chemical shifts ratios between according to R and a, b values, linear correlations (Figure 9) were observed.
| Hatree | ev | kcal/mol | KJ/mol | |
|---|---|---|---|---|
| LUMO | -0.06972 | -189.713 | -437.495 | -183.05 |
| HOMO | -0.21939 | -596.976 | -137.668 | -576.008 |
| A=electron affinity | 0.06972 | 189.713 | 437.495 | 183.05 |
| I=ionization potential | 0.21939 | 596.976 | 137.668 | 576.008 |
| ΔE=energy gap | 0.14967 | 407.263 | 939.184 | 392.959 |
| χ=electronegativity | 0.144555 | 393.344 | 907.087 | 379.529 |
| Pi=chemical potential | -0.144555 | -393.344 | -907.087 | -379.529 |
| ω=electrophilic index | 0.000781882 | 0.02128 | 0.49063 | 205.283 |
| IP=Nucleophilic index | -0.01081777 | -0.29436 | -678.819 | -284.021 |
| S=molecular softness | 133.627 | 363.609 | 8385.15 | 35083.9 |
| η=molecular hardness | 0.074835 | 203.631 | 469.592 | 196.479 |
Table 9: The calculated electronic structure parameters with DFT/B3LYP/631G (d) of the molecule
Figure 9: The ESP of the molecule
Investigation of thermodynamics properties of compound
Thermodynamics parameters of molecule calculated with B3LYP/DFT631G (d) and B3LYP/HF631G (d) basis sets and shown in the Table 10. Thermodynamic parameters of molecule (Table 10) (such as thermal energy, zero-point vibrational energies (ZPVE), heat capacity, entropy, rotational temperatures and rotational constants) were calculated 298.150 K and 1 atm of pressure. In addition to, the standard thermodynamic functions of heat capacity CV0, entropy S0 and enthalpy H0 were obtained at the B3LYP/DFT/HF 631G (d,p) level.
| Rotational temperatures (Kelvin) | DFT | HF |
|---|---|---|
| A | 0.03712 | 0.03764 |
| (B | 0.01158 | 0.0115 |
| C | 0.00922 | 0.00966 |
| Rotational constants (GHZ) | DFT | HF |
| A | 0.77344 | 0.78431 |
| B | 0.24128 | 0.23969 |
| C | 0.19214 | 0.20137 |
| Thermal Energies E(kcal/mol) | DFT | HF |
| Translational | 0.889 | 0.889 |
| Rotational | 0.889 | 0.889 |
| Vibrational | 139.66 | 149.712 |
| Total | 141.437 | 151.489 |
| Thermal Capacity CV(cal/mol-K) | DFT | HF |
| Translational | 2.981 | 2.981 |
| Rotational | 2.981 | 2.981 |
| Vibrational | 52.793 | 48.68 |
| Total | 58.755 | 54.641 |
| Entropy S(cal/mol-K) | DFT | HF |
| Translational | 42.402 | 42.402 |
| Rotational | 33.46 | 33.407 |
| Vibrational | 53.711 | 49.924 |
| Total | 129.574 | 125.733 |
| Zero-point correction (Hartree/Particle) | 0.209677 | 0.226634 |
| Thermal correction to Energy | 0.225394 | 0.241414 |
| Thermal correction to Enthalpy | 0.226339 | 0.242358 |
| Thermal correction to Gibbs Free Energy | 0.164774 | 0.182618 |
| Sum of electronic and zero-point Energies | -869.644549 | -864.521565 |
| Sum of electronic and thermal Energies | -869.628832 | -864.506786 |
| Sum of electronic and thermal Enthalpies | -869.627888 | -864.505841 |
| Sum of electronic and thermal Free Energies | -869.689453 | -864.565581 |
| Zero-point vibrational energy (Kcal/mol) | 131.57455 | 142.21514 |
Table 10: The calculated thermodynamics parameters of the molecule
Conclusion
In this work, geometrical parameters and spectroscopic parameters such as IR, 1H-NMR and 13C-NMR spectra of molecule are calculated by Density Functional Theory (DFT) and Hartree-Fock (HF) methods with the 631G (d) basis sets of the program package Gaussian G09W. Obtained spectroscopic parameters are compared with experimental data. Otherwise, calculated theoretical properties of the compound according to two different basis sets were compared. In the result, the obtained data with B3LYP/HF631G (d) basis sets were found to be closer to the experimental data. The chemical shifts in the calculations 1H-NMR and 13C-NMR and IR vibrational frequencies are found to be compatible with the experimental data. Theoretical and experimental carbon and proton chemical shifts ratios between acording to R2 and a, b values, linear correlation were observed. The possitive frequency in the IR data was found. This result, structures of compound were shown stable. In addition, the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), bond lengths, bond angles, mulliken charges, ELUMO-EHOMO energy gap (ΔEg), electronegativity (χ), electron affinity (A), global hardness (η), softness (σ), ionization potential (I), total energy of the molecule, thermodynamics properties (thermal energies (E), entropy (S), thermal capacity (CV), dipole moments were calculated B3LYP/HF 631G (d) basis sets.
References
- W. Gao, Z. Zheng, Molecules., 2002, 7(7), 511-516.
- M. Shakir, M. Azam, Y. Azim, S. Parveen, A.U. Khan, Polyhedron., 2007, 26(18), 5513 - 5518.
- F. Aydogan, N., Ocal, Z., Turgut, C. Yolacan, Bull. Korean Chem. Soc., 2001, 22(5), 476-480.
- F. Azam, S., Singh, S.L. Khokhra, O. Prakash, J. Zhejiang Univ. Sci., 2007, B8(6), 446-452.
- A. Arslantas, H. Yüksek, O. Gürsoy-Kol, Z. Ocak, Z. Tomruk, M. Calapoglu, Asian J. Chem., 2012, 24(8), 3327-3334.
- O. Gürsoy-Kol, H. Yüksek, F. Islamoglu, Rev. Chim., 2012, 63(11), 1103-1111.
- O. Gürsoy-Kol, H. Yüksek, E. J. Chem., 2010, 7(1), 123-136.
- H. Bayrak, A. Demirbas, S.A. Karaoglu, N. Demirbas, Eur. J. Med. Chem., 2009, 44(3), 1057-1066.
- M. Pitucha, A. Olender, M. Wujec, P. Borowski, M. Mardarowicz, J. Chin. Chem. Soc., 2010, 57(2), 260-265.
- H. Yüksek, A. Demirbas, A.A. Ikizler, C.B. Johansson, C. Çelik, Arzneim Forsch, Drug Res., 1997, 47, 405-409.
- H.A. Saadeh, I.M. Mosleh, A.G. Al-Bakri, M.S. Mubarak, Monatsh. Chem., 2010, 141(4), 471-478.
- M.A. Henen, S.A.A. El Bialy, F.E. Goda, M.N.A. Nasr, H.M. Eisa, Med. Chem. Res., 2012, 21(9), 2368-2378.
- A.A. Ikizler, F. Ucar, H. Yüksek, A. Aytin, I. Yasa, T. Gezer Acta Pol. Pharm. Drug Res., 1997, 54(2), 135-140.
- N. Upmanyu, J.K. Gupta, K. Shah, P. Mishra, J. Pharm. Bioallied Sci., 2011, 3(2), 259-265.
- Z.Y. Li, Y. Cao, P. Zhan, C. Pannecouque, J. Balzarini, E. De Clercq, X.Y. Liu, Lett. Drug Des. Disc., 2013, 10(1), 27-34.
- N. Demirbas, R. Ugurluoglu, A. Demirbas, Bioorg. Med. Chem., 2002, 10(12), 3717-3723.
- C.B. Zhang, W.C. Yang, X.Q. Deng, Z.S. Quan, Med. Chem. Res., 2012, 21(10), 3294-3300.
- N. Chidananda, B. Poojary, V. Sumangala, N.S. Kumari, P. Shetty, T. Arulmoli, Eur. J. Med. Chem., 2012, 51, 124-136.
- K.A. Ali, E.A. Ragab, T.A. Farghaly, M.M. Abdalla, Acta Pol. Pharm., 2011, 68(2), 237-247.
- A.R. Bhat, G.V. Bhat, G.G. Shenoy, J. Pharm. Pharmacol., 2001, 53(2), 267-272.
- G. Turan-Zitouni, Z.A. Kaplancikli, K. Erol, F.S. Kiliç, II, Farmaco, 1999, 54(4), 218-223.
- M. Pitucha, A. Chodkowska, R. Duda, E. Jagiełło-Wojtowiczb, Z. Naturforsch, 2009, 64, 615-618.
- H. Yüksek, F. Akdeniz, Ş. Bahçeci, Synthesis of Some Potentially Biologically active 4,5-dihydro-1H-1,2,4-triazol-5-one Derivatives and investigation of their structure, Chemistry 2006, XX, National Chemistry Congress, Kayseri, Abstract book, 167, 2009.
- M.J. Frisch, Gaussian Inc., Wallingford, CT, 2009.
- R. Dennington, T. Keith, (Edi.), Millam, J. GaussView, Version 5, Semichem Inc., Shawnee Mission KS, 2009.
- K. Wolinski, J.F. Hilton and P.J. Pulay, Am. Chem. Soc., 1990, 112, 512.
- M.H. Jamróz, Vibrational Energy Distribution Analysis: VEDA 4 program, Warsaw, 2004.
- N. Sundaraganesan, S. Ilakiamani, H. Saleema, P.M. Wojciechowski, D. Michalska, Acta A., 2005, 61, 2995-3001.
- C. James, A.A. Raj, R. Reghunanthan, V.S. Jayakumar, I.H. Joe, J. Raman Spectrosc., 2006, 37, 1381-1392.



