Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 1
Identification and Synthesis of New Process Related Impurity in Brimonidine Tartarate
Hemant V Chavan1*, Raju M Patil2, Shriram D Ganapure2, Shivkumar S Jalde1 and Sagar T Sankpal1
1Department of Chemistry, A.S.P. College Devrukh, Dist: Ratnagiri-415 804, Maharashtra, India
2Department of Chemistry, Institute of Science, 15, Madam Cama Road, Mumbai-400 032, India
- Corresponding Author:
- Hemant V Chavan
- Department of Chemistry, A.S.P. College Devrukh Dist: Ratnagiri-415 804, Maharashtra, India
Abstract
In the present report, we identified one process related impurity in brimonidine tartarate its Relative Retention Time (RRT) is 2.23. Liquid Chromatography-Mass Spectroscopy (LC-MS) showed its molecular ion peak as 248. It was isolated by preparative High Performance Liquid Chromatography (HPLC), characterized by Infra-Red (IR), Proton Nuclear Magnetic Resonance (1H-NMR) and mass spectral analysis revealed the unknown impurity as 5-Bromo-quinoxalin-6-yl-cyanamide (7). Further it was confirmed by synthesis starting from quinoxaline thiourea. Both compounds isolated by HPLC and synthesized compound confirms the unknown impurity in brimonidine tartarate as 5-Bromo-quinoxalin- 6-yl-cyanamide (7).
Keywords
Brimonidine, Ophthalmic, Impurity
Introduction
Impurities in pharmaceuticals are the unwanted chemicals that remain with the Active Pharmaceutical Ingredients (API), or develop during formulation or upon aging of both API and formulated APIs to medicine. There is an ever increasing interest in impurities present in API’s. Recently, not only purity profile but also impurity profile has become essential as per various regulatory requirements. The International Conference on Harmonization (ICH) began to publish definitive guidelines on impurities in drug substances and drug products in the late 1990s [1], these guidelines have been adopted by the regulatory bodies, among others, the USA, the European Union and Japan have enjoyed wide support within the pharmaceutical industry over the intervening decade. Under this guidance, the normal qualification threshold for known or identified impurities is 0.15% (1500 Parts Per Million (ppm)) 1 mg/day, whichever is lower, for drug substances whose intake is up to 2g/day. Impurities which exceed this threshold must have their toxicity specifically investigated. Below the qualification threshold, no investigation is required, although impurities at level above 1000 ppm (or 1 mg/day) are expected, at the least, to be identified [2].
Brimonidine is a clonidine derivative that is administered ocularly to lower Intraocular Pressure (IOP) in patients with ocular hypertension or open angle glaucoma [3]. Elevated IOP has long been recognized as a major factor for human glaucoma [4]. A common and effective treatment for glaucoma is the use of IOP lowering topical drugs that acts at a variety of cellular targets, such as the α2 and β adrenergic receptors [5]. Brimonidine is the only selective α-adrenergic receptor agonist approved for chronic treatment in glaucoma and it is indicated for reducing IOP in patients with open angle glaucoma or OHT. Brimonidine is a selective α2-adrenergic receptor agonist that shows upto 1780 fold selectivity for α2 over α1-adrenergic receptor [6].
There are various methods reported for the preparation of brimonidine tartarate. Most of the process as reported use the common intermediate 6- thioisocyanato-quinoxaline (1), which was synthesized directly [7] or generated in situ [8], the former method involves treatment of 5-bromo-6- amino quinoxaline with thiophosgene as shown in Scheme 1 (Method A), latter method synthesized quinoxaline thiourea from 5-bromo-6-amino quinoxaline and benzoyl isocyanate, thiourea is converted into 6-isocyanato-quinoxaline (1). The 6-thioisocyanato-quinoxaline reacts with ethylenediamine to give brimonidine (Scheme 1, Method B).
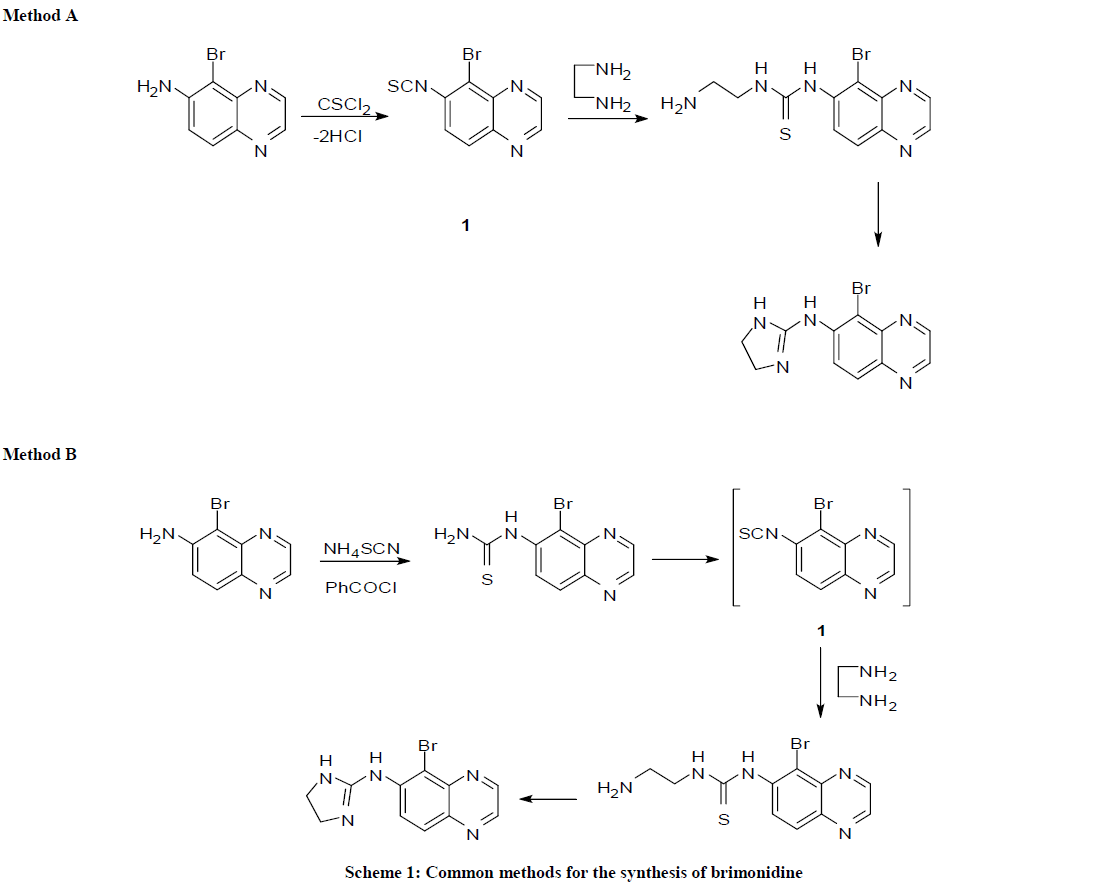
Scheme 1: Legend
In another method brimonidine was synthesized from 5-bromo-6-amino quinoxaline and imidazoline sulfonic acid using triethylamine as base in acetonitrile [9]. Glushkov et al., [10] synthesized the brimonidine from iminium chloride, 6-amino-5-bromo quinoxaline and ethylenediamine. Naik et al., [11] have synthesized the brimonidine using 1-acetylimidazolin-2-one and 6-amino-5-bromo quinoxaline in POCl3. In the literature there is only one paper reported for brimonidine impurities (Compounds 2-4) [12] as shown in Scheme 2.
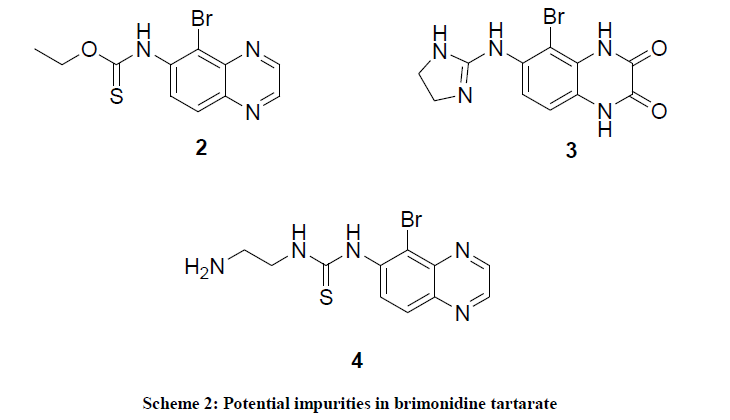
Scheme 2: Potential impurities in brimonidine tartarate
Expermenttal Section
Preparation of cyanamide impurity (7)
A mixture of compound 6 (10 g), iodine (22.26 g), triethylamine (21.41 g) and 150 ml of ethyl acetate was added to the reaction, it was allowed to stir for overnight, the progress of reaction was monitored by HPLC. Reaction mass was concentrated under reduced pressure to get residue, to the residue 100 ml of water was added allowed to stir for 30 min and add 20% of 200 ml of sodium thiosulfate solution was added, allowed to stir for 15 min, reaction mass was filtered. The filtrate was acidified with 1:1 dilute HCl upto 2 pH. Extract with 200 ml of Dichloromethane (DCM) stirred for 15 min, organic layer separated, again 2 × 50 ml of DCM added stirred for 15 min layer separated. Combined organic layer was concentrated under reduced pressure to get the product 7 and purified by column chromatography using DCM as eluent (5 g, 57%).
M.P. 244-249°C (decomp); IR (KBr, cm-1): 3430, 3052, 2948, 2225, 1614, 1496, 1418, 1261, 1042, 977, 876; 1H-NMR (DMSO, 300 MHz): δ=8.99 (d, 1H, J=1.8 Hz), 8.89 (d, 1H, J=1.8 Hz), 8.15 (d, 1H, J=9 Hz), 7.74 (d, 1H, J=9.3 Hz); 13C-NMR (75 MHz, DMSO): 146.80, 144.60, 140.40, 139.44, 138.50, 130.23, 120.06, 111.24, 107.50; Mass: 248 (M+).
Cyanamide impurity (7) isolated from preparative HPLC: IR (KBr, cm-1): 3255, 3149, 3050, 2226, 1662, 1612, 1512, 1488, 1358, 1086, 982, 868, 711; 1H-NMR (DMSO, 400 MHz): δ=8.95 (s, 1H), 8.83 (s, 1H), 8.10 (s, 1H, J=12 Hz), 7.73 (d, 1H, J=12 Hz); Mass: 248 (M+).
Results and Discussion
During synthesis of brimonidine tartarate, an unknown impurity of 12% and its RRT is 2.23. An initial attempt to identify the impurity, it was analyzed form LC-MS. It showed a molecular ion peak at 248. There was difference of 34 mass from the product. This could be elimination of H2S from product and fragmentation also confirms this structure (Scheme 3). The compound was purified using mixture of DMF and Methanol. Even after repeated crystallizations the impurity was not removed, whereas the unknown impurity was enriched from 12% to 30%. Further we tried to isolate the impurity by column chromatography, but isolation of pure impurity was unsuccessful, because of very close RF value of both impurity and product
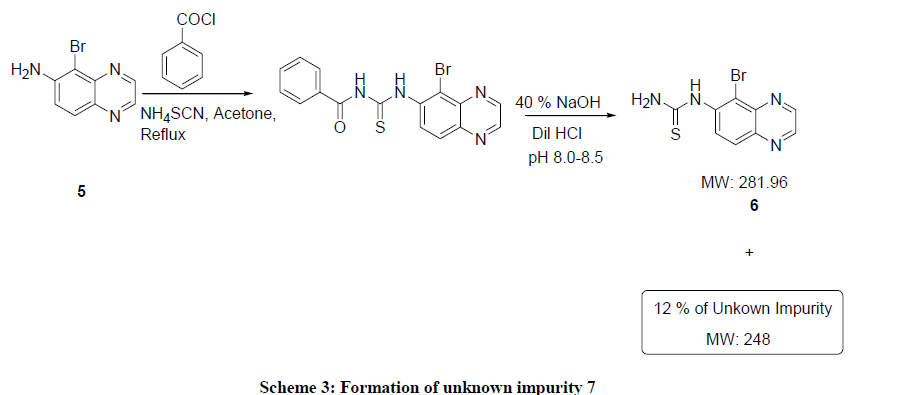
Scheme 3: Formation of unknown impurity 7
As the isolation of impurity by column chromatography was tedious job, next option was to isolate the unknown impurity by preparative HPLC. We isolated 25 mg of unknown impurity by this method and it was characterized it by IR, 1H-NMR and mass spectrometric analysis. All these spectroscopic data confirmed it as cyanamide impurity (7). In the IR spectrum there was a sharp band at 2226 cm-1, which indicates the CN stretching. In 1H-NMR showed the 4 aromatic protons and GC-MS showed the molecular ion peak at 248 which confirms the mass of the 5- Bromo-quinoxalin-6-yl-cyanamide impurity (7).
The unknown impurity was formed during neutralization with dilute HCl. The critical parameter is the pH range it should be 8.0-8.5, below this there could be probability of formation of this impurity. In order to study the effect of pH on the impurity formation, we carried out 3 experiments at 7, 5 and 3 pH respectively and results were shown in the Table 1.
| Experiment No. | pH | Resulta |
|---|---|---|
| 1 | 7 - 8 | 96.19: 3.81 |
| 2 | 5 - 6 | 91.1: 8.9 |
| 3 | 3 - 4 | 85: 15 |
aHPLC purity of compound 6: 7
Table 1: Study of the effect of pH on the impurity formation
From the table it revealed that, slight decrease in pH below 8 (viz., 7-8) results in 4% of impurity formation (Table 1, entry 1), further decrease in pH below 7 (viz., 5-6) gave 9% (Table 1, entry 2) and pH below 5 (viz., 3-4) results in 15% of impurity formation (Table 1, entry 3).
After the identification of impurity structure it was synthesized by synthetic method. There were several methods reported in the literature which includes; cyanation of amines using reagent cyanogen bromide and sodium carbonate [13]. Cyanamides were also prepared from thiourea using Diactoxy Iodobenzene (DIB) [14], iodine and triethylamine [15], iodine, hydrogen peroxide and sodium bicarbonate [15]. Cyanamides were also prepared from deoxygenating of isocyanate [16] using palladium catalysis [17]. Finally, this impurity was synthesized using the method reported by Nath et al., with slight modification [18], starting from quinoxaline thiourea 6, iodine and triethylamine in ethyl acetate at room temperature (Scheme 4). Initially, we obtained the desired product 7 and reactant 6, these two were inseparable in TLC and it was difficult to purify the product. Later we optimized reaction condition to get the cyanamide impurity 7; the results were shown in Table 2.
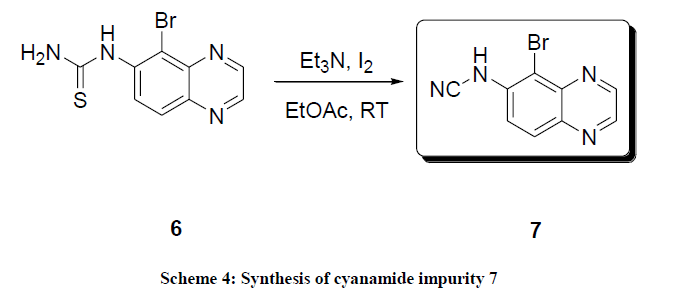
Scheme 4: Synthesis of cyanamide impurity 7
| Experiment No. | Reagents (Iodine: Triethylamine) | Resultsa |
|---|---|---|
| 1 | 1: 4 | 90: 10 |
| 2 | 2: 6 | 96: 4 |
| 3 | 2.5: 6 | 99.3: 0.7 |
aHPLC purity of compound 6 and 7
Table 2: Optimization of reaction condition to synthesize cyanamide impurity
Results depicted in Table 2 shows that, 2.5 and 6 equivalent of iodine and triethylamine required to give product 7 exclusively (Table 2, entry 3). Initially we used 1:4 equivalents of iodine and triethylamine as per reported method [15], only 90% product formation was observed and remaining 10% was reactant 6. In the next experiment both iodine and triethylamine ratio was increased to 2:6 gives 96% of product formation and 4% reactant 6 (Table 2, entry 2).
In order to avoid this impurity formation pH adjustment is very crucial; the pH range should be 8.5-9.5. It is better to adjust the pH with the help of the pH meter by using cooled dilute HCl instead of hot. By controlling these two parameters (pH range and cooled HCl) we can easily avoid the formation of this impurity.
This synthesized cyanamide impurity (7) has melting point 244-249°C (decomp). Its IR spectrum showed the prominent peak at 2225 cm-1 which indicates the presence of cyano group. In 1H-NMR spectrum there were 4 aromatic protons. In 13C-NMR absence of signal of carbon attached to sulfur at 180 ppm indicates the absence of thiourea linkage and peak at 107 ppm corresponded to nitrile (CN) carbon which confirmed the structure of cyanamide impurity. GCMS showed molecular ion peak at 248 also confirmed the structure of cyanamide impurity. Both the compounds isolated by semi-preparative HPLC and synthesized cyanamide compound confirmed the unknown impurity was 5-Bromoquinoxaline- 6-yl-cynamide (7).
References
- ICH guideline: Impurities in New Drug Substances Q3A (R2), International Conference on Harmonization 2006; ICH Guideline: Impurities in New Drug Products Q3B (R2), International Conference on Harmonization, 2006.
- A recent FDA guideline on impurities in generic drugs would effectively reduce the qualification threshold to 1000 ppm or 1 mg/day for new impurities which were not present in the reference listed drugs 2009.
- D. Cambridge, Eur. J. Pharmacol., 1981, 72, 413-415.
- J.C. Morrison, J. Glaucoma., 2005, 14, 315-17.
- J.C. Tsai, E.M. Kanner, Expert. Opin. Emerg. Drugs., 2005, 10(1), 109-118.
- L.B. Cantor, Expert. Opin. Pharmacother., 2000, 1, 815-834.
- J.C. Danielewicz, M. Snarey, G.N. Thomas, Patent US-3890319 A, 1975.
- M.B. Thomos, M.T. Williams, United Kingdom, Patent-1463520(A), 1977.
- S.A. Munk, D. Harcourt, P. Arasasingham, C. Gluchowski, H. Wong, J. Burke, A. Kharlamb, C. Manlapaz, E. Padillo, L. Williams, L. Wheeler, M. Gargt, Bioorg. Med. Chem. Lett., 1995, 5(15), 1745-1748.
- R.G. Glushkov, A.I. L'vov, L.N. Dronova, G.A. Modnikova, I.E. Mamaeva, RU-2285003 C1, 2006.
- A.M. Naik, S.D. Sawant, G.A. Kavishwar, S.G. Kavishwar, Int. J. Pharm. Tech., 2010, 2, 14-17.
- L. Kvapil, M. Grepl, P. Hradil, Acta Universitatis Palackianae Olomucensis Facultas Rerum Naturalium., 2003, 42, 19-26.
- C.C. Lindsey, B.M. O’Boyle, S.J. Mercede, T.R.R. Pettus, Tetrahedron Lett., 2004, 45, 867-868.
- H. Ghosh, R. Yella, A.R. Ali, S.K. Sahoo, B.K. Patel, Tetrahedron Lett., 2009, 50, 2407-2410.
- L. Jamir, U.B. Sinha, J. Nath, B.K. Patel, Synth. Comm., 2012, 42, 951-958.
- F.F. Wong, C.Y. Chen, M.Y. Yeh, Syn. Lett., 2006, 559-562.
- M.H. Larraufie, G. Maestri, M. Malacria, C. Ollivier, L. Fensterbank, E. Lacote, Synthesis., 2012, 44, 1279-1292.
- J. Nath, B.K. Patel, L. Jamir, U.B. Sinha, K.V.V.V. Satyanarayana, Green Chem., 2009, 11, 1503-1506.



