Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 4
Hirshfeld Computational Analysis and Crystal Structure of 5-[3-(4-Nitrophenyl)-4,5-dihydro-1,2-oxazol-5-yl]-Methyl-5H-dibenzo-[b,f]azepine
Manjunath BC1, Madan Kumar S2, Lingaraju GS3,4, Sadashiva MP3, Lokanath NK5* and Byrappa K4
1Department of Physics, Yuvaraja College, University of Mysore, Mysore, India
2DST-PURSE Lab, Mangalore University, Mangalore, India
3Department of Studies in Chemistry, University of Mysore, Mysore, India
4Department of Materials Science, Mangalore University, Mangalore, India
5Department of Studies in Physics, University of Mysore, Mysore, India
- *Corresponding Author:
- Lokanath NK
Department of Studies in Physics
University of Mysore
Mysore, India
Abstract
The title compound 5-[3-(4-Nitrophenyl)-4,5-dihydro-1,2-oxazol-5-yl]Methyl-5H-dibenzo-[b,f]azepine, is synthesized and characterized using single crystal X-ray diffraction method. The azepine ring adopts boat confirmation and the fused benzene rings to the azepine ring makes a dihedral angle of 53.39 (7)0. In the crystal structure, molecules are stabilized with the C-H...π, N-O...π and π...π short contacts. The intermolecular contacts in the crystal structures are quantified using Hirshfeld surfaces analysis method. The results suggests that the intermolecular H...H contacts contributes more to the crystal packing when compared to other contacts and the electrostatic potential surfaces are visualized.
Keywords
Dibenzoazepine, Crystal structure, Short contacts, Hirshfeld surfaces
Introduction
A large number of dibenzoazepine derivatives have been synthesized and reported to have number of pharmacological activities [1-3]. Among them, dibenzo[b,f]azepine have attracted considerable attention, which shows a broad range of pharmaceutically active compounds [3-5]. Dibenzoazepine deravative, 5H-dibenzo[b,f]azepine is a common basic fused tricyclic amine. It is used as an intermediate for the synthesis of the registered anticonvulsant drug oxcarbazepine [6,7]. Dibenzo[b,f]azepine and its derivatives have been variously reported as having antiallergic activity, specifically antihistaminic, spasmolytic, serotonin antagonistic, anticonvulsive, antiemetic, antiepileptic, antiinflammatory, sedative and fungicidal action [8]. The crystallographic studies of the molecular and crystal structures of dibenzoazepine derivatives have been reported. [9-12]. In view of the above importance, the synthesis, crystal structure and Hirshfeld surfaces of the title compound 5-[3-(4- Nitrophenyl)-4, 5-dihydro-1,2-oxazol-5-yl]Methyl-5H-dibenzo-[b,f]azepine is reported herewith.
Materials and Methods
Synthesis of the 5-[3-(4-Nitrophenyl)-4,5-dihydro-1,2-oxazol-5-yl]-Methyl-5H-dibenzo-[b,f]azepine
4-Nitro-benzaldehyde oxime (2.0 mmol) was taken in methylene dichloride (10 ml) taken in a round bottom flask. The solution was made basic using triethyl amine (5.0 mmol). N-Chloro succinimide (2.0 mmol), followed by tricyclic amine (1 mmol), were added slowly at 0°C to the stirring solution. After complete addition of the reactants the stirring was continued 6 h. After completion of the reaction (Monitored by Thin Layer Chromatography (TLC)), the reaction mixture was extracted with ethyl acetate (2 × 25 ml). The combined organic layer was washed with 0.1 N hydrochloric acid (2 × 20 ml), followed by brine solution (2 × 20 ml). Then, the organic layer was dried over anhydrous sodium sulphate, filtered and concentrated under reduced pressure to afford the crude product, which was then purified by column chromatography over silica gel (60-120 mesh) using Hexane: Ethyl acetate mixture in 7: 3 ratios as eluent. The slow evaporation method using ethyl acetate and hexane gave yellow colored block shaped crystals. The schematic diagram of the molecule is shown in Scheme 1.
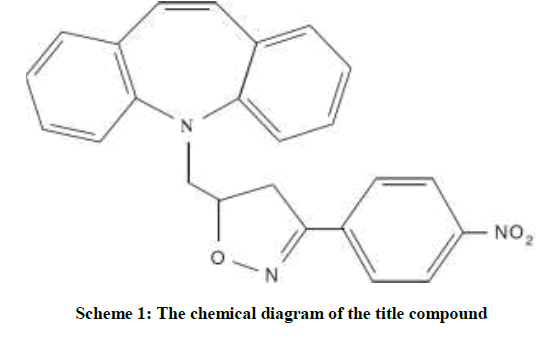
Scheme 1: The chemical diagram of the title compound
Single crystal X-ray diffraction
A yellow block shaped crystal of the title compound, C24H19N3O3 with dimensions of 0.23 × 0.21 × 0.22 mm3 was chosen for an X-ray diffraction study using Bruker X8 Proteum diffractometer equipped with CuKα radiation (λ =1.54178 Å) at 296K. Data collections, integration, and scaling of the reflections were performed using the APEX2 program [13]. The structure was solved by direct methods using SHELXS [14]. All the non-hydrogen atoms were revealed in the first Fourier map itself. Full-matrix least squares refinement using SHELXL [14] with isotropic displacement parameters for all the atoms Refinement of the non-hydrogen atoms with anisotropic parameters was started at this stage. The hydrogen atoms were placed at chemically acceptable positions and were allowed to ride on their parent atoms. The details of the crystal data and structure refinement are given in Table 1. The ORTEP [15] of the molecule with thermal ellipsoids drawn at 50% probability is shown in Figures 1 and 2.
| CCDC number | 1526804 |
|---|---|
| Empirical formula | C24H19N3O3 |
| Formula weight | 397.42 |
| Temperature (K) | 293 (2) |
| Wavelength (Kα,Å) | 0.71075 |
| Crystal system, space group | Monoclinic, P21/c |
| Unit cell dimensions (Å, º) | a=18.8298 (11) b=6.9338 (4) c=15.1055 (10) β=95.096 (2) |
| Volume Å3 | 1964.4 (2) |
| Z, Calculated density (Mg/m3) |
4, 1.344 |
| Absorption coefficient (mm-1) | 0.733 |
| F (000) | 832 |
| Crystal size mm | 0.20 x 0.20 x 0.21 |
| Theta range for data collection (0) | 2.4 to 63.8 |
| Limiting indices | -21 ≤ h ≤ 21, -8 ≤ k ≤ 6, -17 ≤ l ≤ 14 |
| Reflections collected/unique[R (int)] | 21670/3091 [0.030] |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 3091/0/371 |
| R value/wR2 | 0.0390/0.1094 |
| Goodness-of-fit on F2 | 1.04 |
| Largest diff. peak and hole (e. Å-3) | 0.26 and −0.31 |
Table 1: Crystal data and structure refinement details for the title compound
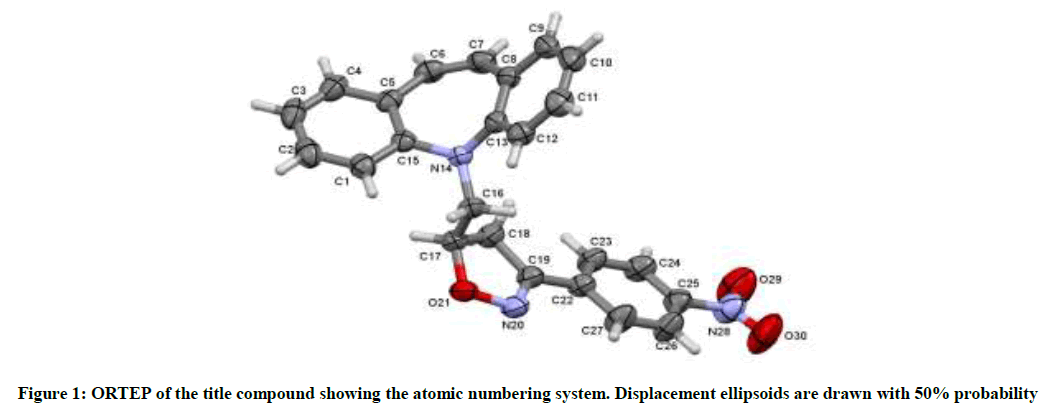
Figure 1: ORTEP of the title compound showing the atomic numbering system. Displacement ellipsoids are drawn with 50% probability
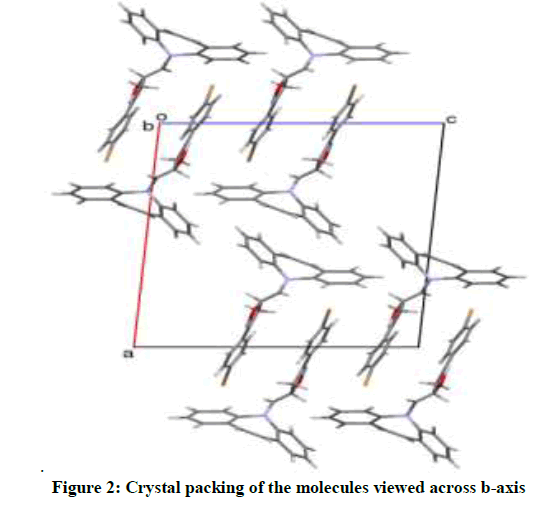
Figure 2: Crystal packing of the molecules viewed across b-axis
Hirshfeld surfaces analysis
The program Crystal Explorer 3.0 [16] was used to perform Hirshfeld surfaces computational analysis and to quantify the intermolecular interactions in terms of surface contribution and generating graphical representations (Figure 3a), plotting 2D fingerprint plots (Figure 3b) [17] and generating electrostatic potential (3c) [18] with TONTO [19]. The electrostatic potential is mapped on Hirshfeld surfaces using Hartree-Fock (STO-3G basis set) theory over the range of −0.020 a.u. to + 0.020 a.u. (Figure 3c).The electrostatic potential surfaces are plotted with red region which is a negative electrostatic potential (hydrogen acceptors) and blue region which is a positive electrostatic potential (Hydrogen donor).

Figure 3: Hirshfeld surfaces analysis: (a) dnorm mapped on Hirshfeld surface for visualizing the intercontacts of the title compound. Color scale in between −0.003 au (blue) and 1.400 au (red). (b) Fingerprint plots- (i)C...C, (ii) C...H, (iii) C...N, (iv) C...O, (v)H...H (vi)H...N, (vii)H...O and (viii) full. The outline of the full fingerprint is shown in gray. This is the closest internal distance from a given point on the Hirshfeld surface and de is the closest external contacts. (c) Electrostatic potential mapped on Hirshfeld surface (different orientation) with ± 0.020 au. Blue region corresponds to positive electrostatic potential and red region to negative electrostatic potential
Results and Discussions
Molecular and crystal structure
The azepine ring (Figure 1) adopts a boat conformation with the nitrogen atom showing a maximum deviation of 0.494(1) Å, the puckering parameters Q2=0.711(1) Å, φ2=1.95(1)◦, Q3=0.214(1) Å and φ3=359.8(4)◦ and the total puckering amplitude QT=0.7431(1) Å [20]. The fused benzene rings Cg(2):C1/C2/C3/C4/C5/C15 and Cg(3) C8/C9/C10/C11/C12/C13 to the azepine motif makes a dihedral angle of 53.39(7)◦. The bond lengths and bond angles of the title compound are in good agreement with the standard values. The bond lengths N14–C13=1.434(2) Å and N14–C15=1.434(2) Å indicates π−electron delocalization, as a result there is a difference in the bond length of C5–C6=1.459(2) Å and C7–C8 = 1.455(2) Å.6,7. The torsion angles for the atoms C1-C15-N14-C16 and C12-C13-N14-C16 shows +syn-periplanar conformation with 24.8(19)◦ and -syn-periplanar conformation with -27.9(1)◦ about the mean plane of fused rings respectively. The packing of the molecules (Figure 2) is stabilized with the π...π, C-H...π and N-O...π short contacts. The π...π interaction exists between centroids Cg(2) with a distance of 3.927(9) Å [−x, y − 1, −z]. The C3-H3...Cg(3) and C9-H9...Cg(2) exists with distance of 3.643(2) Å [angle 142◦, (−x, y − 1,−z)] and 3.573(2) Å [angle 147◦, (x, y − 1/2, z − 1/2)], respectively. In addition a short contact N28--O29...Cg(3) exists with a distance of 3.737(2) Å [angle 102◦, (x, y − 1/2, z − 1/2)].
Hirshfeld surface analysis
The short contacts (π...π, C-H...π and N-O...π) in the crystal structures are quantified using Hirshfeld surface analysis and the Hirshfield surfaces are visualized (Figure 3a). The 2D-fingerprint plots of intercontacts contributing to the Hirshfeld surfaces are (i) C...C-4.2%, (ii) C...H-19.6%, (iii) C...N-2.9%, (iv) C...O – 4.7%, (v) H...H-43.8%, (vi) H...N -4.1%, (vii) H...O-20.6% and the full plots is shown in Figure 3b, (viii) The major intercontacts contributing to Hirshfeld surfaces are H...H, O...H and C...H. The electrostatic potential Figure 3c shows the distribution of positive and negative potential over the Hirshfeld surfaces.
Conclusions
The title compound is synthesized, crystallized and the three dimensional molecular structure is determined using single crystal X-ray diffraction method. The azepine ring adopts boat configuration and the crystal structure is stabilized with short contacts π...π, C-H...π and N-O...π. In addition, the intercontacts C...C, C...H. C...N, C...O, H...H, H...N and H...O are contributing to the Hirshfeld surfaces.
Acknowledgments
Authors thank IOE, Vignayna Bhavan and University of Mysore for providing single crystal X-ray diffraction and DST-PURSE Lab, Mangalore University.
Supplementary materials
Crystallographic data for the structure reported in this paper have been deposited with the Cambridge Crystallographic Data Center as supplementary publications numbers 1526804.
References
- H. Vijay Kumar, C.R. Gnanendra, N. Nagaraja, D. Channe Gowda, Eur. J. Chem., 2008, 5(S2), 1123-1132.
- P. Pigeon, B. Decroix, Tetrahedron Lett., 1997, 38, 1036-1041.
- G. Steiner, A. Franke, E. Haedicke, D. Lenke, H. Teschendorf, Hofmann, H. Krieskott, W. Worstmann, J. Med. Chem., 1986, 29, 1877.
- Loiseau P., Duche P., Levy R. H., Mattson., Meldrum M. S., In antiepilepticdrugs. Ed. Raven Press, New York, 555-566 (1995).
- D.A. Learmont, J. Benes, A. Parada, D. Hainzl, A. Beliaev, J.B. Maria, M.M. Pedro, A.C. Maria, G. Jose, S. Patricio, Eur. J. Med. Chem., 2001, 36, 227-236.
- M. Lucarini, P. Pedrielli, G.F. Pedulli, L. Valgimigli, D. Gigmes, P. Tordo, J. Am. Chem. Soc., 1999, 121(49), 11546-11553.
- L.J. Krichka, A. Ledwith, Chem. Rev., 1974, 74, 101-123.
- A. Hempel, N. Camerman, A. Camerman, D. Mastropaolo, Acta Crysttallogr., E., 2005, 61, 1313-1315.
- M.M.M. Abdoh, S. Madan Kumar, K.S. Vinay kumar, B.C. Manjunath, M.P. Sadashiva, N.K. Lokanath, Acta Crystallogr. E., 2013, 69, o17.
- B.C. Manjunath, K.S. Vinay Kumar, S. Madan Kumar, M.P. Sadashiva, N.K. Lokanath, Acta Crystallogr. E., 2013, 69, o1233.
- S. Madan Kumar, B.C. Manjunath, G.S. Lingaraju, M.M.M. Abdoh, M.P. Sadashiva, N.K. Lokanath, Crystal Structure Theory and Applications, 2013, 3, 124-131.
- S. Madan Kumar, B.C. Manjunath, K.S. Vinay Kumar, K.J. Pampa, M.P. Sadashiva, N.K. Lokanath, J. Crystallogr., 2014, 1-9.
- Apex2 (Version 2013), Program for Bruker CCD X-Ray Diffractometer Control, Bruker AXS Inc., Madison, Wis, USA, 2013.
- G.M. Sheldrick, Acta Crystallogr. A, 2008, 64(1), 112-122.
- C.F. Macrae, I.J. Bruno, J.A. Chisholm, P.R. Edging-ton, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek, P.A. Wood, J. Appl. Crystallogr., 2008, 41(2), 466-470.
- S.K. Wolff, D.J. Grimwood, J.J. McKinnon, D. Jayatilaka, M.A. Spackman, Crystal Explorer 3.0, University of Western Australia, Perth, Australia, 2001.
- M.A. Spackman, J.J. McKinnon, Cryst. Eng. Comm., 2002, 4(66), 378-392.
- M.A. Spackman, J.J. McKinnon, D. Jayatilaka, Cryst. Eng. Comm., 2008, 10(4), 377-388.
- D. Jayatilaka, D.J. Grimwood, A. Lee A. Lemay, A.J. Russel, C. Taylo, S.K. Wolff, TONTO-A System for Computational Chemistry, The University of Western Australia, Nedlands, Australia, 2005.
- D. Cremer, J.A. Pople, J. Ame. Chem. Soc., 1975, 97(6), 1354-1358.



