Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 10
Effect of an Indole-containing Pseudopeptide on Behavioral Despair Models in Mice: Supporting by Molecular Docking and Density Functional Theory Calculations
Golnaz Mohebzadeh1,2,3, Nahid Fakhraei2, Keivan Akhtari4, Golnoush Mohebzadeh5, Saeed Balalaie6, Keyumars Hassanzadeh7 and Ahmad Reza Dehpour2,3*
1School of Pharmacy, Tehran University of Medical Sciences, International Campus (TUMS- IC), Tehran, Iran
2Brain and Spinal Cord Injury Research Center, Neuroscience Institute, Tehran University of Medical Sciences, Tehran, Iran
3Experimental Medicine Research Center, School of Medicine, Tehran University of Medical Sciences, Tehran, Iran
4Department of Physics, University of Kurdistan, P.O. Box 416, Sanandaj, Iran
5School of Medicine and Surgery, Bologna University of Medical Science, Bologna, Italia
6Peptide research Center, Chemistry Faculty, K N Toosi University of Technology, Tehran, Iran
7Young Researchers and Elites Club, Sanandaj Branch, Islamic Azad University, Sanandaj, Iran
- *Corresponding Author:
- Ahmad Reza Dehpour
Brain and Spinal Cord Injury Research Center
Neuroscience Institute
Tehran University of Medical Sciences
Tehran, Iran
Abstract
Aims: The study refers to application of a synthetic candidate containing an indole scaffold, 5f, in animal models of behavioral despair. Moreover, binding affinity and polarizability of 5f were calculated with molecular docking and density functional theory (DFT) study, respectively.
Materials and Methods: To compare, desipramine (DMI) (10 mg/kg, i.p.), a selective serotonin-norepinephrine reuptake inhibitor (SNRI) and fluoxetine (FLX) (10 mg/kg, i.p.), a selective serotonin reuptake inhibitor (SSRI) were employed. The binding affinity of 5f, FLX and DMI to LeuTa, a leucine tranporter as a homologous protein to monoamine, were evaluated. Also, polarizability of 5f was compared using DFT method. The neuroprotective effect of 5f on behavioral despair was evaluated using forced-swim and tail suspension tests in male mice. The drugs were injected intraperitoneally (i.p.) 30 minutes before the tests.
Results: 5f (2.5, 5, 10 mg/kg) significantly reduced the immobility behavior in a dose-dependent manner in the behavioral tests. Moreover, no significant changes in locomotor activity of the animals were detected. In addition, in line with the experimental results, docking demonstrated that 5f-LeuTAa complex has the highest bonding energy comparable with DMI. Moreover, DFT study showed the highest polarizability for 5f. Further, it showed a high lipophilicity. 5f had higher potency in the behavioral tests compared to the antidepressants; however, it was more structurally similar to DMI. 5f was probably capable to reduce behavioral despair lowering the immobility figure.
Conclusion: Demonstrating a high polarizability and having the highest bonding energy of docked 5f-LeuTAa complex comparable with DMI along, 5f probably has the potential to make more serotonin and norepinephrine available.
Keywords
Indole-containing pseudopeptide, Sodium symporter (NSS) transporter, Forced-swim test, Tail suspension test, Molecular docking, Density functional theory.
Introduction
Intracellular communication in the central nervous system (CNS) is driven by the controlled release and reuptake of neurotransmitters at the synapses. Extra accumulation or lack of monoamine and amino acid neurotransmitters contributes to many psychiatric and neurological disorders, including Parkinson’s disease, Attention Deficiency Hyperactivity Disorder (ADHD) and depression [1,2].
The extent and duration of the released neurotransmitters are tightly regulated by a group of specialized transmembrane neurotransmitter sodium symporter (NSS) transporters localized to the neurons and glial cells. The NSS transporters use the Na+/Cl- electrochemical gradient to remove neurotransmitters from the synapse against their concentration gradient [1,2]. NSS is to terminate neurotransmission by rapid removal of neurotransmitters from the synaptic cleft and reuptake them into the presynaptic neurons and/or surrounding glials. The subfamily of NSS responsible for reuptake of monoamines comprises the transporters for dopamine (DAT), noradrenaline (NET), and serotonin (SERT). The 3-dimensional (3D) structure of SERT is not known, however; several 3D structures of a bacterial homologous transporter, the Aquifex aeolicus leucine transporter (LeuTAa), are available. The first LeuT data revealed a high-resolution occluded structure devoid of water with a single leucine molecule in coordination with two Na+ ions [3]. A remarkable step forward in understanding structure–function of SLC6 NSS transporters came from the Gouaux laboratory with the determination of the high resolution crystal structure of LeuTAa, a prokaryotic member of the SLC6 NSS family [3]. Given that the structural fold of proteins from the same protein family is generally highly conserved [4], it is not surprising that LeuTAa has become the established template for studying both prokaryotic and eukaryotic SLC6 transporters, despite sharing only 20%-25% protein sequence identity [5]. The substrate leucine and two Na+ and one Cl- ions were also observed in the crystal structure. Out of 22 residues forming the substrate binding site, 18 were direct H bond, ionic or hydrophobic interactions with leucine, and the remaining four coordinated one of the Na+ ions (Na1) [3]. Models which were constructed from LeuTAa are now allowing a reasonable approach to further clarify the molecular determinants of NSS transporter–ligand complexes, and potentially the ability to better manipulate drug specificity and affinity. LeuTAa was also used as template to understand how antidepressants bind to SERT and NET. In fact, the low-affinity interaction sufficed to allow visualizing tricyclic antidepressants (TCAs) in the extracellular vestibule of the transporter [6,7]. TCAs such as desipramine and imipramine, binds to serotonin (5-HT) and norepinephrine (NE) transporters with affinities of nanomolar to tens of nanomolar concentrations and blocks transport activity [8].
Indole nucleus is found in numerous natural products and pharmaceuticals. It has found that indole compounds, derivatives and complexes have the potential to be used to assist in treatment of human immune and neurological disorders [9-11]. Indole has gained prominence in medicinal chemistry due to its diverse biological properties [12-14]. Many indole derivatives are considered as the most potent scavenger of free radicals [15]. Indoles have analgesic, antipyretic and anti-inflammatory characteristics. They are essential to maintain healthy functioning of the nervous and immune systems. They are often used in synthesis of new pharmaceuticals [9]. Among the large family of indoles, indole carboxamides have recently attracted a great deal of interest from chemists and biologists due largely to their various pharmacological activities such as antioxidant [16], hypocholestrolemic [17], antihistaminic [18] etc. [19-21]. Although functionalized indole ring systems have been found frequently in biologically active compounds, indole derivatives as multi-component reaction (MCR) partners and flexible reactions for the rapid generation of complex molecules with often biologically relevant scaffold structures [22-24], are rather under-represented [25]. The synthesized indole-containing pseudopeptide possesses optimal range of lipophilicity for oral absorption, cell membrane penetration, and blood-brain barrier (BBB) permeation [26].
Peptidomimetics typically are small protein-like molecules designed to mimic natural peptides or proteins. These mimetics whose structures were mainly derived from natural peptides, should have the ability binding to their natural targets in the same way of the natural sequences and hence produce the same biological impacts. It is possible to design these molecules in such a way that they show the same biological influences as their peptide role models, nevertheless, with enhanced properties like higher proteolytic stability and bioavailability and often with improved selectivity or potency [27]. The purpose is to replace as much of the peptide backbone as possible with non-peptide fragments while still maintaining the pharmacophoric groups, usually the amino acid side chains, of the peptide. This makes the compound more lipophilic, and as a result its bioavailability is improved. Replacement of the amide bond with alternative groups prevents proteolysis and promotes metabolic stability [28].
The discovery of new effective drugs still has remained a top priority as depression affects approximately 21% of the world population. Extensive studies have measured antidepressant-like effects of chemical compounds to discover modern drugs [29]. Clinically-used antidepressants have several limitations and drawbacks which demand persistent development of novel, efficient, and safe drugs. Particular attention has been devoted to synthesis of tricyclic structures as potential scaffolds for novel pseudopeptides [30].
Due to the biological importance of the indole nucleus and its presence in the CNS drugs and the similarity between molecular structure of general anti-depression drugs (Figure 1a) and our synthetic compound (Figure 1b), we aim to demonstrate protective effect of a synthesized novel indole-containing pseudopeptide (5f) in mice exposed to inescapable situations to evaluate behavioral despair in animal models. We compare our result with a TCA, desipramine (DMI), and a SSIR, fluoxetine (FLX). Moreover, to compare capability of 5f with DMI and FLX in possible inhibition of 5-HT or NE-reuptake, we model different types of interactions associated with LeuTAa and the drugs, using molecular docking density functional theory (DFT) method.
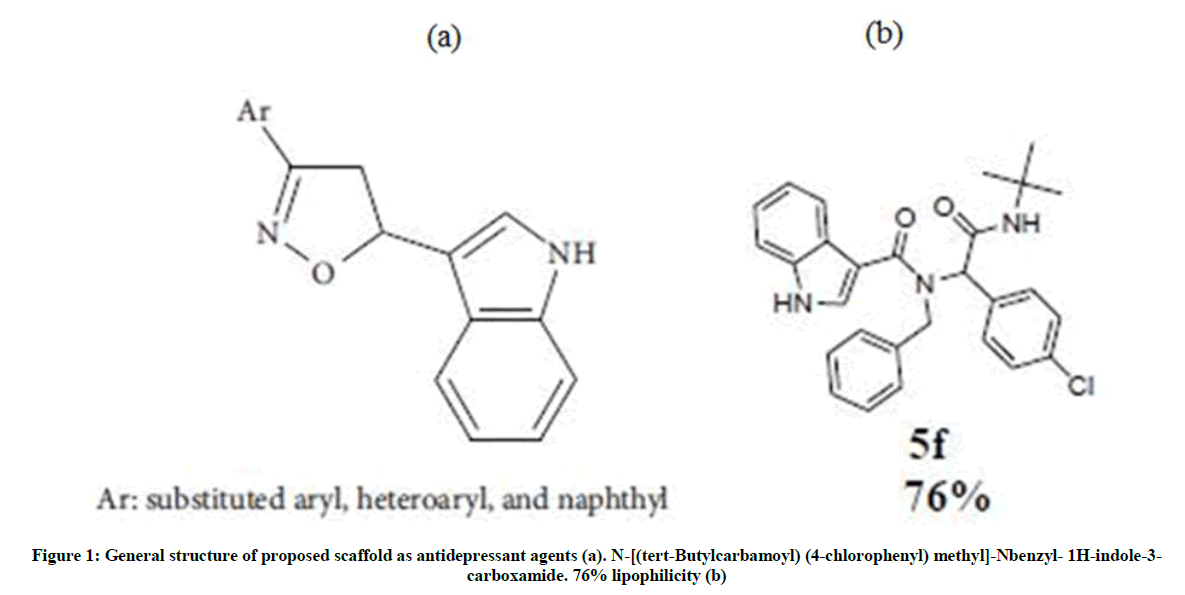
Figure 1: General structure of proposed scaffold as antidepressant agents (a). N-[(tert-Butylcarbamoyl) (4-chlorophenyl) methyl]-Nbenzyl- 1H-indole-3-carboxamide. 76% lipophilicity (b)
Materials and Methods
A novel indole-containing pseudopeptide, 5f, (synthesized in Chemistry Department, Khaje Nasir Toosi University, Tehran, Iran). Desipramine, selective serotonin-norepinephrine reuptake inhibitors (SNRI) and fluoxetine, a selective serotonin reuptake inhibitor (SSRI). Tween 80 (0.5%), paraffin, and glycerin were all purchased from Sigma co.
Animals
Male NMRI mice weighting 23-30 g (Pasteur Institute) were used throughout the study. Animals were allowed free access to food and water. All behavioral experiments were conducted with normal room light (12 h regular light/dark cycle) and temperature (22°C ± 1°C). All the procedures were carried out in accordance with the Institutional Animal Care and Use Committee guidelines for animal care and use. This study was approved by the Ethics Committee of Tehran University of Medical Sciences.
Experimental groups
The animals were divided into 14 groups of eight. Seven groups were assigned for FST and seven similar groups for TST. Three groups were injected intraperitoneally (i.p.) with the synthetic compound, 5f, (2.5, 5, 10 mg/kg). One other group received the compound vehicle [Tween 80 (0.5%), distilled water, paraffin]. Two groups received FLX and DMI as reference drugs. One group received saline as the antidepressant vehicle.
The pseudopeptide was already synthesized by our colleagues in Peptide Chemistry Research Center, K. N. Toosi University of Technology, Tehran, Iran using an Ugi-4CR strategy via the reaction of benzaldehyde derivative, primary amine, indolecarboxylic acid, and isocyanide, which selectively precipitated the desired products out of the reaction mixtures (Table 1) [26]. Derivatives with an electron donating group on the phenyl ring of indolyl pyrazoline appear to be a more promising structural feature than with an electron withdrawing group for antidepressant like effects [31].
| Product | Isocyanide | Acid | Amine | Aldehyde |
|---|---|---|---|---|
| 5f | t-Bu | Indole-3 | PhCH2 | 4-Cl-C6H4 |
Table 1: Synthesized indole-containing pseudopeptide via an Ugi four-component reaction
Chemistry of the synthesized drug, 5f
(5f): N-[(tert-butylcarbamoyl) (4-chlorophenyl) methyl]-N-benzyl-1H-indole-3-carboxamide
M. p. 132ºC-134ºC; IR (KBr, cm-1) υ=3208, 1672 1597; 1H-NMR (500 MHz, CDCl3): δ (ppm)=1.26 (s, 9H, t-Bu), 4.65 (d, 1H, J= 16.25 Hz, CHBenzyl), 4.88 (d, 1H, J=18.2 Hz, CHBenzyl), 5.52 (brs, 1H, NH), 6.16 (s, 1H, CH),7.11-7.30 (m, 13H, HAr), 7.97 (d, 1H, J=7.7, HAr), 9.53 (brs, 1H, NHindole); 13C-NMR (125 MHz, CDCl3): δ (ppm)=28.5, 51.6, 65.7, 110.3, 111.8, 120.7, 121.4, 123.0, 126.4, 126.5, 127.1, 127.2, 128.6, 128.7, 130.7, 134.1, 134.2, 135.8, 137.8, 168.9, 169.5; HR-ESI MS calc. for C28H29ClN3O2 [M+1] + 474.19428, found 474.19427).
Preparation of the drug for animal administration
Due to high lipophilicity of the synthesized compound and impossibility solving in saline, we managed to prepare our compound in suspension form. The compound was levigated with glycerin and then Tween 80 (0.5%) employed as a surfactant and emulsifier. After that, water was gradually added when the solution was under a stirrer. The suspension was used in short-term so that we did not require including stability studies.
Behavioral procedures
Open-field test (OFT)
In order to detect any possible association of immobility in the tests and changes in motor activity, and ensure that alterations in the duration of immobility are not resultant from the changes that occur in motor activity, the locomotor behavior of mice was assessed in an open-field test. The apparatus consisted of a wooden box measuring 40 × 60 × 50 cm. The floor of the cube was divided into 12 equal squares. The animals were gently placed in the left corner of the field and the number of squares crossed with all paws counted. The floor of the open-field apparatus was cleaned between the tests [32,33].
Forced swimming test (FST)
The test was conducted using the method [34] with minor modifications. Mice were individually placed in glass cylinders (20 cm height × 14 cm diameter) containing 15 cm depth of water at 25ºC ± 1ºC for 6 min. Duration of immobility was regarded as the time spent by the mouse floating in the water without struggling and making only those movements necessary to keep its head above the water. The duration of immobility was scored during the last 4 min of the 6-min test period. Each mouse was judged immobile when it ceased struggling and remained floating motionless in the water. Conventional antidepressants decrease the immobility time in this test [35].
Tail suspension test (TST)
The total duration of immobility induced by tail suspension was measured according to the method described [35]. Briefly, mice both acoustically and visually isolated were suspended 50 cm above the floor by adhesive tape placed approximately 1 cm from the tip of the tail. Immobility time was recorded during a 6 min period [36,37].
Molecular docking and density functional theory calculations
To evaluate capability of 5f in possible inhibition or deceleration of 5-HT and or NE-reuptake and compare it with DMI and FLX, different types of interactions between 5f, DMI and FLX were modeled with LeuTAa as a template separately using molecular docking and density functional theory (DFT) methods. Molecular docking plays an important role in molecular biology study and drug discovery. In our calculations, molecular docking has been performed using HEX 6.3 software [38]. To study the binding affinity of 5f to LeuTAa, the X-ray crystallographic 3D structure of LeuTAa complex with imipramine (PDB id 2Q72) [7] was downloaded from the online Protein Data Bank RCSB PDB (http://www.pdb.org).
After removing imipramine molecules, the molecular docking has been performed with 5f. Similar simulation has been performed with a LeuTAa and two standard references FLX and DMI.
Visualization of the docked pose has been done using CHIMERA- (www.cgl.ucsf.edu/chimera) and PyMOL (http://pymol.sourceforge.net/) tools.
To give a deeper insight to the higher binding affinity of 5f with respect to FLX and DMI, DFT study was employed. Geometry optimization and single point energy calculations were carried out using DFT method supplemented in the GAUSSIAN9 8W suite of the programs [39], Becke3-Parameter hybrid functional combined with the Lee–Yang–Parr correlation functional, abbreviated as B3LYP [40,41,42] with the standard 6-31G* basis set.
The polarizability of a molecule measures the relative tendency of its electronic cloud to be distorted from its normal shape by a weak external electric field [43] and can be considered as a global reactivity descriptor [44]. To further investigate the compounds reactivity descriptors, the polarizability tensor elements of 5f, FLX and DMI were computed at B3LYP/6-31G* level of theory. The mean values of polarizabilities for the studied molecules are calculated using the following equation:
α0= (αXX + αYY + αZZ)/3 (1)
Where αXX, αYY and αZZ are diagonal components of the tensor of polarizability. The mean values of polarizabilities are 327.596, 197.052, and 188.043 au for 5f, DMI and FLX, respectively. In theoretical point of view, it can be easily concluded that 5f has more reactivity advantages with respect to FLX and DMI due to the highest polarizability.
Statistical analysis
The data was analyzed using SPSS software (Version 18). The data were expressed as mean ± SEM. The one-way ANOVA was run, followed by Tukey’s post hoc comparisons test. The criterion for the statistical significance was P<0.05.
Results
The resulting binding energies of docked 5f-LeuTAa, FLX-LeuTAa and DMI-LeuTAa complexes were found to be -315.11, -174.42 and -229.75 kJ/mol, respectively. These values show the highest binding affinity to LeuTAa for 5f and the lowest for FLX.
The docked complexes of 5f/LeuTAa are shown in Figure 2. 5f ligand with surrounding active site residues within 3.5 Å and the spatial orientation in binding pocket are given in Figure 3.
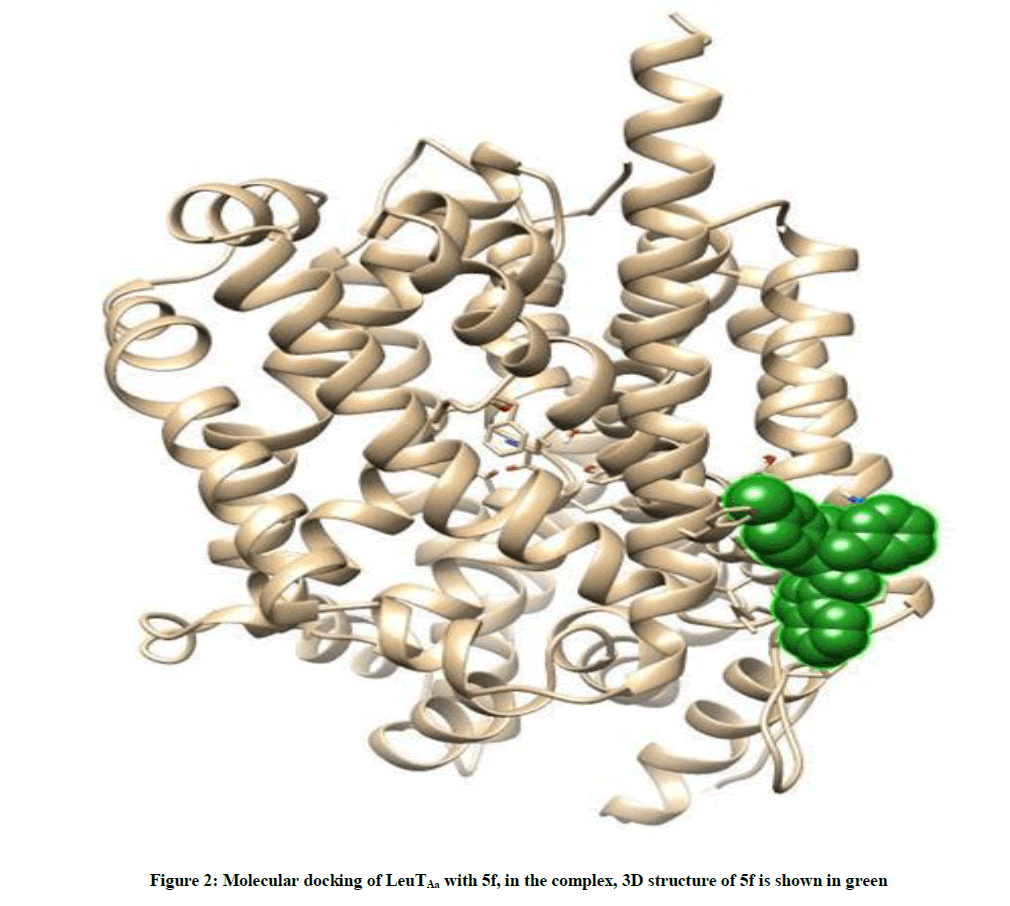
Figure 2: Molecular docking of LeuTAa with 5f, in the complex, 3D structure of 5f is shown in green
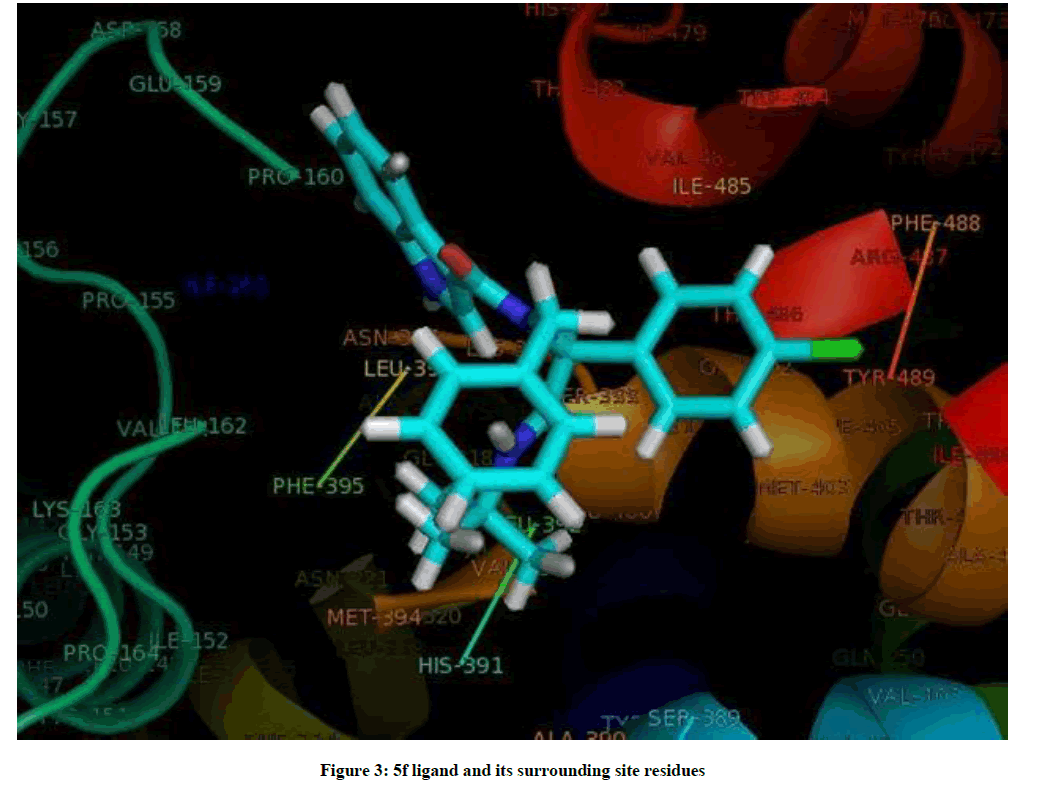
Figure 3: 5f ligand and its surrounding site residues
Three-dimensional molecular electrostatic potential (MEP) surfaces for 5f, FLX and DMI are depicted in Figure 4. The negative electronic potential is shown in red, while the positive electrostatic potential is shown in blue, according to the color code ranges, considering the code numbers, 5f can act more effectively in charge-controlled interactions compared to the standard drugs.
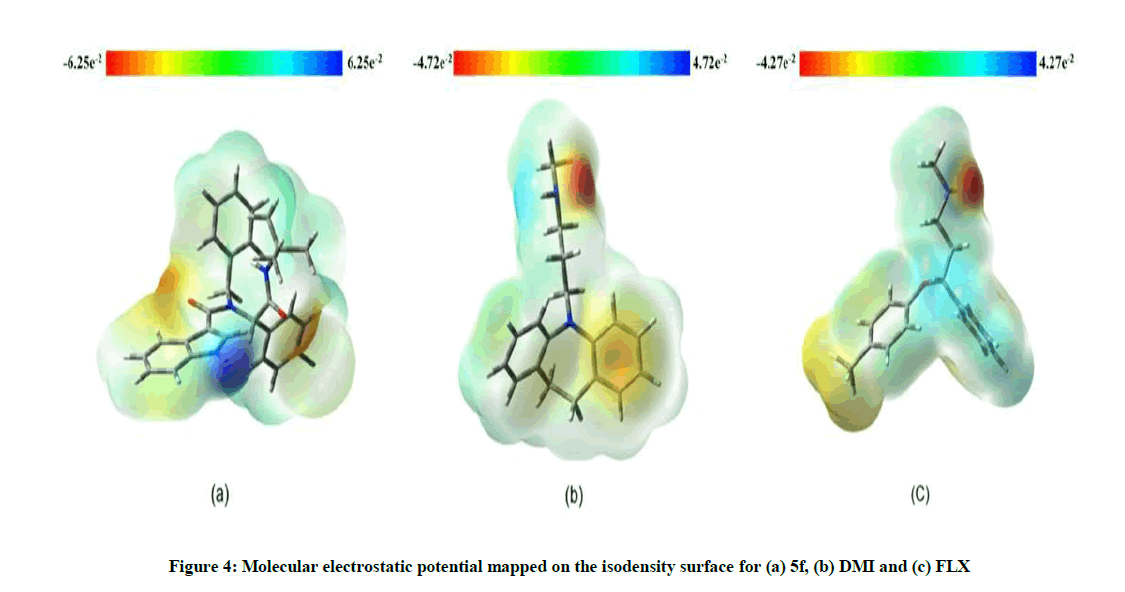
Figure 4: Molecular electrostatic potential mapped on the isodensity surface for (a) 5f, (b) DMI and (c) FLX
As can be seen in Figure 5, the drug 5f dose dependently (2.5, 5, 10 mg/kg) lowered the immobility time in FST and this effect was significant in doses 5 and 10 mg/kg, P<0.001, P<0.001, respectively. The result was comparable to the reference drugs, FLX (20 mg/kg) and DMI (5 mg/kg) which markedly lowered the immobility measures, P<0.001 and P<0.001, respectively (Figure 5a). Also, locomotor activity of the mice did not influence following drugs administration (Figure 5b).
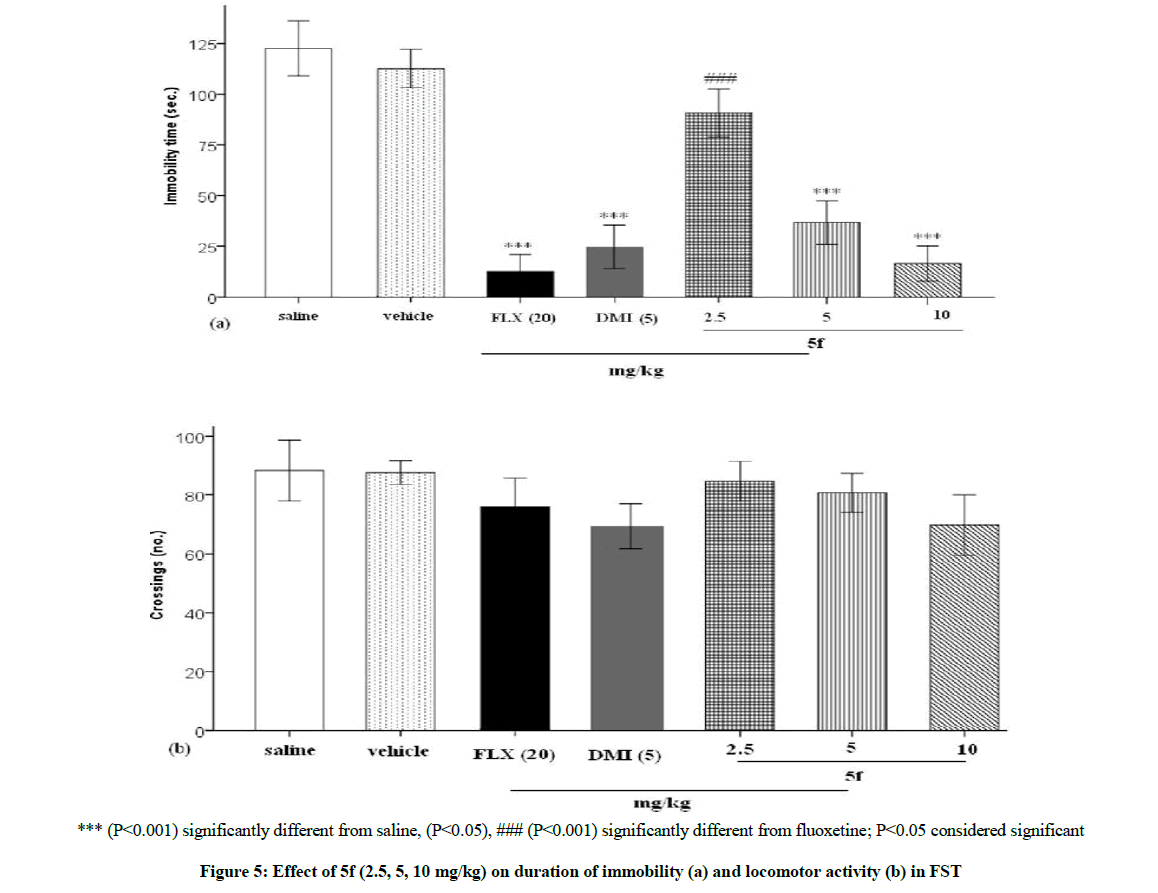
*** (P<0.001) significantly different from saline, (P<0.05), ### (P<0.001) significantly different from fluoxetine; P<0.05 considered significant
Figure 5: Effect of 5f (2.5, 5, 10 mg/kg) on duration of immobility (a) and locomotor activity (b) in FST
Figure 6 represents the results of TST and it is clear that 5f dose dependently (2.5, 5, 10 mg/kg) reduced the immobility figure. The doses 2.5, 5, 10 mg/kg markedly lowered the immobility time, P<0.01, P<0.01, P<0.001, respectively. Furthermore, the result of the drug in dose 10 mg/kg was comparable with FLX (20 mg/kg) and DMI (5 mg/kg) which markedly lowered the immobility measures, P<0.001 and P<0.001, respectively.
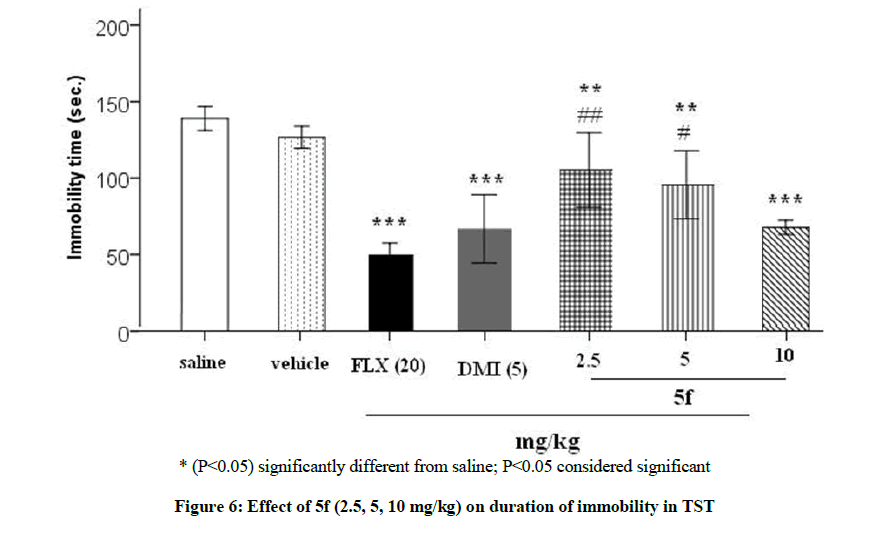
* (P<0.05) significantly different from saline; P<0.05 considered significant
Figure 6: Effect of 5f (2.5, 5, 10 mg/kg) on duration of immobility in TST
Discussion
The results of this investigation revealed a biological activity for the pseudopeptide 5f and it demonstrated protective central effect in the behavioral despair tests comparable with DMI. Further, molecular docking and DFT study also proved for the first time that 5f is more structurally similar to DMI and also forms a significantly more powerful complex with LeuTAa than the standards. Docked 5f-LeuTAa showed the highest binding energy in comparison with the other complexes, however; this figure was much more close to docked DMI- LeuTAa. On the other hand, 5f indicated significantly higher polarizability compared to the standard drugs in DFT study which demonstrated that 5f complex is profoundly stronger.
Several studies have shown antidepressant potential of indole derivatives [45,46]. Similarly numerous indole derivatives are reported to exhibit potent anticonvulsant activity [47,48]. Marine indole alkaloids were mentioned as potential new drug leads for control of depression and anxiety [49].
Antidepressant activity of some new 5-(1H-indol-3-yl)-3-(substituted aryl)-4, 5-dihydroisoxazoline derivatives was evaluated and it clearly demonstrated that replacement of aromatic core by appropriate heterocycles such as pyridine and pyrrole on the 5-(1H-indol-3-yl)-3-(Phenyl)-4, 5-dihydroisoxazoline (3a) would generate more potent derivatives. Thus, these compounds can serve as potential leads for further antidepressant studies [31].
Notably, amino acid tryptophan having indole moiety is the precursor of neurotransmitter serotonin and melatoin. Other indolic compounds include plant hormone auxin, indomethacin (anti-inflammatory drug), pindolol (beta blocker agent) etc. [50]. Indole derivatives have been shown in various studies that acted in CNS disorders such as epilepsy, depression and anxiety [51].
Bali et al. used phenyl hydrazine and substituted acetophenones as starting materials to synthesize novel indole derivatives. These derivatives were then screened for their atypical antipsychotic profile using apomorphine-induced mesh climbing method [52]. Replacement of phenyl ring by heterocyclic nucleus on the proposed scaffold increases antidepressant activity and in our experiment replacement had been done by pyrrole binding of various tricyclic and other types of antidepressants to LeuT using a scintillation proximity assay has been carried out [53]. Consistent with our study, several tricyclic compounds (imipramine, nortriptyline, protriptyline, amitriptyline and doxepin) showed binding affinity, however; desipramine bound LeuT most tightly, with an IC50 of 80 μM. Desipramine binds at the inner end of the extracellular cavity of the transporter and is held in place by a hairpin loop and by a salt bridge. This binding site is separated from the leucine-binding site by the extracellular gate of the transporter. By directly locking the gate, desipramine prevents conformational changes and blocks substrate transport. In addition, fluoxetine also showed measurable binding. Desipramine was found to inhibit leucine binding to LeuT by decreasing its maximal binding capacity without changing its binding affinity, indicating a mechanism that does not involve competitive inhibition [6]. TCA-binding site is probably conserved from bacterial to mammalian NSS proteins. Therefore, in the human Na+/Cl--dependent neurotransmitter transporters SERT, NET and DAT it is likely that TCAs as well as SSRIs also bind between the extracellular gate and EL4 hairpin, thereby inhibiting neurotransmitter reuptake at the synapse. In combination with homology modeling [54] and molecular docking, the identification of this drug-binding site will allow studies on interactions of SERT-, NET- and DAT-specific inhibitors [55] with these transporters and may aid in the structure-based design of more effective neurotransmitter reuptake inhibitors as antidepressants [6].
Moreover, our indole-containing pseudopeptide, sharing structural similarities with 5-HT, it might have a competitive nature with 5-HT in binding to its transporter (SERT) and by which enhances 5HT in the presynaptic membrane. Further, the possibility remains to be understood that if 5f also binds to noradrenaline transporter (NET), as well as SERT. As a result, 5f has the potential to probably facilitate the understanding of 5-HT and NE receptor functions and perhaps generate new drug leads for the treatment of depression, anxiety, migraines and other cognitive disorders.
Conclusion
Due to high lipophilicity, 5f probably could pass the blood brain barrier (BBB). Furthermore, presumably it diminished the behavioral disappointment forming a stronger complex with LeuTAa. This structure, however; may indicate possible affinity to LeuTAa. Considering binding energies of the drug/LeuTAa complexes and the mean polarizabilities of the studied drugs, the DFT-B3LYP calculations are in a good agreement with the experimental results. However, our synthetic drug 5f proved to be more structurally similar to DMI. Moreover, as 5f showed higher potency in our behavioral tests compared to the standard drugs, it may form a significantly more powerful complex. Nevertheless, further study will be needed to further extend these results and show the exact mechanisms. It should make clear that what subfamily of NSS in reuptake of monoamines is probably responsible and which transporters, dopamine (DAT), noradrenaline (NET), and serotonin (SERT), and which monoamines are really involved. Moreover, it is required to examine our synthetic compound in other central experiments related to cognition in different animal models.
Acknowledgement
We would like to thank Nahid Sadeghi Alavijeh in Chemistry Department, Khaje Nasir Toosi University, Tehran, Iran for synthesis and providing the pseudopeptide.
References
- J. Masson, C. Sagné, M. Hamon, S. El Mestikawy. Pharmacol. Rev., 1999, 51, 439.
- N.H. Chen, M.E.A. Reith, M.W. Quick, Pflugers Archiv., 2004, 447, 519.
- A. Yamashita, S.K. Singh, T. Kawate, Y. Jin, E. Gouaux, Nature., 2005, 437, 215-223.
- C. Chotia, A. Lesk, EMBO J., 1986, 5, 823-826.
- S.F. Altschul, T.L. Madden, A.A. Schäffer, J. Zhang, Z. Zhang, W. Miller, D.J. Lipman, Nucleic Acids Res.,1997, 25(17), 3389-3402.
- Z. Zhou, J. Zhen, N.K. Karpowich, R.M. Goetz, C.J. Law, M.E. Reith, D.N. Wang Leu, Science., 2007, 317(5843), 1390-1393.
- S.K. Singh, A. Yamashita, E. Gouaux, Nature., 2007, 448(7156), 952-956.
- A.J. Eshleman, M. Carmolli, M. Cumbay, C.R. Martens, K.A. Neve, A. Janowsky, J. Pharmacol Exp Ther., 1999, 289(2), 877-885.
- E. Lodyga-Chruscinska, M. Turek, Pharma. Chem., 2009, 8, 6.
- A. Ahmad, W.A. Sakr, K.M. Rahman, Cancers (Basel)., 2011, 3, 2955-74.
- A. Katsman, K. Umezawa, B. Bonavida, Curr. Pharm. Des., 2009, 15, 792-808.
- S.S. Panda, P.V.R. Chaudhari, Ind. J. Pharm. Sci., 2008, 70, 208-215.
- K. Bajaj, V.K. Srivastava, S. Lata, R. Chandra, A. Kumar, Indian J. Chem., 2003, 42B, 1723-1728.
- J.S. Biradar, B.S. Sasidhar, R. Parveen, Eur. J. Med. Chem., 2010, 45, 4074-4078.
- Y.J. Chyan, B. Poeggler, R.A. Omar, D.G. Chain, B. Frangione, J. Ghiso, M.A. Pappolla, J. Biol. Chem., 1999, 274 21937e21942.
- H.Y. Aboul-Enein, I. Kruk, K. Lichszteld, T. Michalska, A. Kiadna, S. Marczynski, S. Olgen, Luminescence., 2004, 19, 1-7.
- R. Bellemin, J. Decerprit, D. Festal, Eur. J. Med. Chem., 1996, 31, 123-132.
- S. Battaglia, E. Boldrini, F.D. Settimo, G. Dondio, C.L. Motta, A.M. Marini, G. Primofiore, Eur. J. Med. Chem., 1999, 34, 93-105.
- V. Grumel, J.Y. Merour, B. Lesur, T. Giboulot, A. Frydman, G. Guillaumet, Eur. J. Med. Chem., 2002, 37, 45-62.
- M. Kitano, A. Kojima, K. Nakano, A. Miyagishi, T. Noguchi, N. Ohashi, Chem. Pharm. Bull., 1999, 47, 1538-1548.
- D. Carbonnelle, M. Duflos, P. Marchand, C. Chauvet, J.Y. Petit, F. Lang, J. Pharmacal. Exper. Ther.,2009, 331, 710-716.
- P. Slobbe, E. Ruijter, R.V.A. Orru, Med. Chem. Commun., 2012, 3, 1189-1218.
- A. Domling, W. Wang, K. Wang, Chem. Rev., 2012, 112, 3083-3135.
- I. Akritopoulou-Zanze, Curr. Opin. Chem. Biol., 2008, 12, 324-331.
- J. Sapi, J.Y. Laronze, Arkivoc., 2004, 7, 208-222.
- N.S. Alavijeh, S. Ramezanpour, M.S. Alavijeh, S. Balalaie, F. Rominger, M. Anil Bijanzadeh, Monatsh. Chem., 2013, 2, 349-356.
- A. Grauer, B. Konig, Eur. J. Org. Chem., 2009, 30, 5099-5111.
- R.B. Silverman, In The Organic Chemistry of Drug Design and Drug Action; Elsevier Academic Press: Evanston, IL, USA, 2004, 47-50.
- J.F. Cryan, M.E. Page, I. Lucki, Psychopharmacol. (Berl)., 2005, 182, 335-44.
- I. Cerminara, L. Chiummiento, M. Funicello, A. Guarnaccio, P. Lupattelli, Pharmaceuticals., 2012, 5, 297-316.
- P.O. Patil, S.B. Bari, J. Chem., 2013a, 1-7.
- M.P. Kaster, P.K. Ferreira, A.R. Santos, A.L. Rodrigues, Behav. Brain. Res., 2005, 165, 204-209.
- M. Khoshnoodi, N. Fakhraei, A.R. Dehpour, Pharm. Biol., 2015, 53, 739-745.
- R.D. Porsolt, A. Bertin, M. Jalfre, Arch. Int. Pharmacodyn. Ther., 1977, 229, 327-336.
- L. Steru, R. Chermat, B. Thierry, P. Simon, Psychopharmacol. (Berl)., 1985, 85, 367-370.
- A.L.S. Rodrigues, G.L. Silva, A.S. Matteussi, E.S. Fernandes, O.G. Miguel, R.A. Yunes, J.B. Calixto, A.R. Santos, Life Sci., 2002, 70, 1347-1358.
- M. Mantovani, R. Pértile, J.B. Calixto, A.R. Santos, A.L. Rodrigues, Neurosci. Lett., 2003, 343, 1-4.
- D.W. Ritchie, V. Venkatraman, Ultra-fast FFT protein docking on graphics processors. Bioinformatics., 2010, 26, 2398-2405.
- M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, V.G. Zakrzewski, J.A. Montgomery, GAUSSIAN 98, RevisionA.7, Gaussian, Inc. Pittsburgh PA, 1998.
- C. Lee, W. Yang, R.G. Parr, Phys. Rev., 1988, B37, 785.
- A.D. Becke, J. Chem. Phys., 1993, 98, 5648.
- P.J. Stevens, F.J. Devlin, C.F. Chabalowski, M.J. Frisch, J. Phys. Chem., 1994, 98, 11623-11627.
- H. Kurtz, D. Dudis, Rev. Comput. Chem.,Wiley-VCH, New York, 1998.
- K. Akhtari, K. Hassanzadeh, B. Fakhraei, N. Fakhraei, H. Hassanzadeh, S.A. Zarei, Comp. Theor. Chem., 2013, 1013, 123-129.
- T. Varvaresou, A. Siatra-Papastaikoudi, A. Tsotinis, Tsantili-Kakoulidou, A. Vamvakides, Farmaco., 1998, 53, 320-326.
- P.O. Patil, S.B. Bari, Bioorg. Med. Chem., 2013b, 21, 2434-2450.
- L. Singh, M.J. Field, C.A. Vass, J. Hughes, G.N. Woodruff, Br. J. Pharmacol., 1992, 105, 8-10.
- V. Sharma, P. Kumar, D.J. Pathak, Heterocyclic Chem., 2010, 47 491-502.
- A.J. Kochanowska-Karamyan, M.T. Hamann, Chem. Rev., 2010, 110, 4489-4497.
- R. Dhani, A. Avinash, S.K. Salenaagina, M.V.S. Teja, P. Masthanaiah, P.R. Rathnam, V.C. J. Silpa, Chem. Pharm. Res., 2011, 3, 519-523.
- T. Saini, S. Kumar, B. Narasimhan, Cent. Nerv. Syst. Agents Med. Chem., 2016, 16, 19-28.
- A. Bali, U. Sen, T. Peshin, Eur. J. Med. Chem., 2014, 74, 477-90.
- M. Quick, J.A. Javitch, Proc. Natl. Acad. Sci. USA. 2007, 104, 3603-3608.
- L.R. Forrest, A. Tavoulari, Y.W. Zhang, G. Rudnick, B. Honig, Proc. Natl. Acad. Sci. USA., 2007, 104, 12761-12766.
- B. Olivier, W. Soudijn, I. van Wijngaarden, Prog. Drug Res., 2000, 54, 59-119.



