Research Article - Der Pharma Chemica ( 2023) Volume 15, Issue 2
Design and Synthesis of some 1,3,4-Thiadiazol Amines: Molecular Docking, In-Silico Adme and Antioxidant Properties
Satyanarayan ND*, Sakshith Prasad R and Harishkumar SSatyanarayan ND, Department of Pharmaceutical Chemistry, Kuvempu University, Karnataka, India, Email: satya1782005@gmail.com
Received: 04-Feb-2023, Manuscript No. dpc-20-19225; Editor assigned: 06-Feb-2023, Pre QC No. dpc-20-19225; Reviewed: 20-Feb-2023, QC No. dpc-20-19225; Revised: 21-Feb-2023, Manuscript No. dpc-20-19225; Published: 28-Feb-2023, DOI: 10.4172/0975-413X.15.2.35-43
Abstract
Oxidative stress and associated diseases arising from the overproduction of free radicals can be counteracted by designing and creating novel antioxidative agents that can defend the human body against the damage caused by free radicals. In present study, 5-[2-(thiophen-2-yl)quinolin-4-yl]-1,3,4-thiadiazol-2-amine and its derivatives 5(a-e) were designed and synthesised using the 2-(thiophen-2-yl)quinoline-4-carboxylic acid and thiosemicarbazide. Designed targeted antioxidant compounds were docked against the protein PDB-ID: 3VB8 and evaluated the antioxidant activity using the DPPH assay. The in-silico pharmacokinetic parameter study, such as ADME of the synthesized compounds, was evaluated using AdmetSAR software. The synthesized molecules 5(a-e) shows acceptable values.
Keywords
Quinoline; Thiophene; DPPH assay; Admet 90SAR; 3VP8
INTRODUCTION
Antioxidants are a health-protecting component in food. Antioxidants are trapped in free radicals. Free radicals are molecules or atoms having an unpaired electron and are extremely unstable, capable of taking part in rapid change reactions that destabilise other molecules and create even more free radicals. Free radicals of oxygen play a significant role in the aetiology of various conditions like arthritis, cancer, atherosclerosis, etc. Oxidative DNA damage can play a crucial role in ageing. Where oxidation occurs during aerobic respiration, free radicals are produced in the human body. The oxygen-based free radicals are Reactive Oxygen Species (ROS). These occur in different types, including superoxides (O2•-), peroxyl (ROO•), alkoxyl (RO•), hydroxyls (HO•), and nitric oxides (NO•). Free radicals also derive from other sources such as cigarette smoking, air pollution, ultraviolet rays and ionising radiation. These free radicals cause premature ageing and wrinkles as well as cancer and other chronic illnesses. The free radicals may oxidise nucleic acids, proteins and lipids or DNA that could cause degenerative diseases. Oxygen is required to all aerobic organism's survival, but when supplied at higher concentrations, it may become toxic. Oxygen is fairly non-reactive in its ground state and its partial reduction results in Active Oxygen Species (AOS) such as hydrogen peroxides, singlet oxygen, superoxide radical anion etc. This is partly due to the oxidative stress, which is essentially the oxidant's detrimental effect on physiological activity. Oxidation is a process of removing one or more electrons from an atom or molecule, which results in the formation of free radicals. Such free radicals are deactivated by antioxidants in plants and animals. These antioxidants serve as an inhibitor in the oxidation phase, even at a relatively low concentration and hence have a varied physiological role in the body. Antioxidants are the major protection mechanism for the body which acts as free radical scavengers. In the body, antioxidants include enzymes such as dismutase, catalase, cytochrome P450, peroxidase and glutathione. Carotenoids are found in fruit and vegetables; vitamin C and vitamin E, phenolic compounds, anthocyanine. Included as food additives are butylated hydroxyanisole and hydroxytoluene and tertbutylhydroquinone. All antioxidants have a similar role in inhibiting free radicals, regardless of antioxidant sources. The human body has three levels of antioxidant capacity. The first is that the enzymes are working to regulate initial free development of radical substances. Superoxide, hydrogen peroxide and hydroxyl radicals are all typically formed when oxygen is taken in and used during aerobic breathing. The production of hydroxyl radicals is decreased by catalase and dismutase enzymes. Both peroxides, including hydrogen peroxides, have been eliminated. The co-factors of metal are required by all enzymes. For example, copper, zinc or manganese are needed, depending on pH [1 -7].
The second class is the dietary antioxidants. In many fruits and vegetables, including sweet potatoes, dietary antioxidants are found. Dietary antioxidants are important in the human body because they are able to end the free radical chain reaction by contributing protons. Replenishers are the third group of antioxidants. The source of protons comes from structures which, though being stable, can easily give a proton so that it is not free radical. Carotenoids, flavonoids coenzyme Q and glutathione are examples of replenishers. Free radical-induced oxidative damage is considered as a major risk factor for many diseases and ageing. Free radical is an atom or molecule with one or more unpaired electrons. These are highly unstable, highly reactive and generated in the body constantly during metabolic reactions. Free radicals are also formed during phagocytises and from toxic environmental pollutants, ionising radiation, heavy metal poisoning, cigarette smoke, alcohol. Free radicals being highly reactive can oxidise biomolecules leading to tissue injury and cell death. These are normally removed by enzymatic and non-enzymatic antioxidant defence mechanisms. Deficiency of such mechanisms leads to oxidative damage. To prevent such damage antioxidant preparations are widely used by many people all over the world. An antioxidant is a molecule which inhibits other molecule's oxidation. Oxidation is a chemical reaction transferring electrons or hydrogen to the oxidising agent from a material. Although existence depends on oxidation reactions, they can also lead to damage [8-11].
Antioxidant activity of thiodiazole
The alkaloid dendrodoa grossularite, which is derived from the marine algae dendrodoa, is an alkaloid (5-[(3-N-dimethylamino)-1, 2, 4-thiadiazoly]- 3-indanyl methanone). It comprises a 1, 2, 4-thiadiazole assembly, which is unusual in nature. Some 5-[(2-(phenyl substituted)-1H-benzimidazole-1-yl)methyl]-N-methyl-1, 3, 4- thiadiazole-2-amines have been synthesised and proved by different in-vitro systems for the purposes of antioxidantresearch. A slightly inhibited lipid peroxide in 10 M-3 M concentrations, the compound that represents the most active derivative [12].
MATERIALS AND METHODS
Chemicals used in compound synthesis has been purchased from Alfa Aesar and Spectrochem Pvt. Uh, Ltd. Solvents were of reagent quality and if needed, distilled and dried using routine methods. The Melting Points (M.Pt.) of the synthesised compounds were measured using the digital Raga digital melting point apparatus and are not corrected; the infrared data was reported on the Bruker spectrophotometer using KBr pellets. 1H and 13C NMR spectra were reported on Bruker AVANCE II 400 and 100 MHz devices using DMSO-d6/CDCl3 as solvent and TMS as internal standard; chemical shifts are expressed as d values (ppm). The values of J are expressed in Hertz (Hz). Mass Spectra (MS) were used in the JEOL GCMATE II LC-Mass spectrometer using the Electron Impact ionisation (EI) technique. Analytical Thin-Layer Chromatography (TLC) was performed on pre- coated TLC sheets of silica gel 60 F254 (Merck, Darmstadt, Germany), visualised by long and short wavelength UV lamps. Chromatographic purification with Merck silica gel (100 mesh to 200 mesh) was performed.
Molecular docking
Preparations of ligands were carried out by Chimera docking tool here the energy minimization and merging of non-polar hydrogen done with this tool. 3D structure of protein was downloaded by protein data bank online server PDB ID: 3VB8 Resolution: 2.64 Å and minimization of energy and application of force field carried out by Chimera docking tool. Selection of Binding pocket to hit ligands grid was constructed center_x = -35.3690, center_y = -3.7928, center_z = -7.4265, size_x = 33.4045, size_y = 57.7908, size_z = 394264. Visualization of docked ligand and target protein interaction was carried out using Discovery studies 2020.
General procedure for synthesis of 2-(thiophen-2-yl)quinoline-4-carboxylic acid and its derivatives 3(a-e)
2-acetyl thiophene (0.01 mol) and Isatin (0.01 mol) is taken in a round bottom flask add ethanol and catalytic amount of 33% KOH solution. Thereaction mixture was refluxed at 75°C for 8 hrs. The reaction mixture is observed by TLC to know the status of the reaction. After the completion ofreaction, that is kept out in room temperature. Then pour the reaction mixture drop wise with stirring into the crushed ice and neutralized by usingdilute HCl. The solid separated is filtered and dried in room temperature [13].
Synthesis of 5-[2-(thiophen-2-yl)quinolin-4-yl]-1,3,4-thiadiazol-2-amine and its Derivatives 5(a-e)
2-(thiophen-2-yl)quinoline-4-carboxylic acid (0.01 mol) and Thio semi carbazide (0.01 mol) is taken in a round bottom flask add catalytic amount of POCl3 acid. The reaction mixture is kept for stirring at temperature 100℃ to 110℃. The reaction mixture is observed by TLC to know the status of the reaction. After the completion of reaction, that is kept out in room temperature. Then pour the reaction mixture drop wise with stirring into the crushed ice and neutralized by using dilute KOH. The solid separated is filtered and dried in room temperature.
5-[2-(thiophen-2-yl)quinolin-4-yl]-1,3,4-thiadiazol-2-amine (5a)
Mp: 142℃ to 146℃; Yield: 67%; IR(KBr: υmax/cm-1): C=C(1640cm-1), 3358(C-NH), 1594(C=N), 3500-3300cm-1(N-H), 1690(N=C), 2550(S- H); 1H NMR (400 MHz) δ (ppm): 5.795(S, 2H), 7.587(T, 2H, J=7.6Hz), 7.768(T, 2H, J=7.6Hz), 7.921(D, 1H, J=8Hz), 8.031 (D, 2H, J=8.4Hz), 8.652 (S,1H). 13C NMR (400 MHz) δ (ppm): 103.1, 120.1, 125.5, 127.0, 127.6, 127.9, 128.3, 129.0, 129.9, 140.0, 144.9, 147.6, 158.1, 161.6, 175.0. MS: m/z=309.99 (M+1).
5-[6-chloro-2-(thiophen-2-yl)quinolin-4-yl]-1,3,4-thiadiazol-2-amine (5b)
Mp: 148℃ to 152℃; Yield: 72%; IR(KBr: υmax/cm-1): C=C(1640cm-1), 3358(C-NH), 1594(C=N),3500-3300cm-1(N-H), 1690(N=C), 785-540(C- Cl); 1H NMR (400 MHz) δ (ppm): 7.178(T,1H,J=4.4Hz), 7.687(S,1H), 7.726(D,1H,J=4.4Hz), 7.859(D,1H,J=2.8Hz), 8.0465(D,1H, J=4.4Hz), 8.139(S,1H), 8317(S, 1H), 8.501(D, 1H, J=2.4Hz). 13C NMR (400 MHz) δ (ppm): 122.834, 124.556, 124.556, 124.819, 125.402, 127.942, 128.477,128.798, 129.148, 130.860, 131.094, 131.580, 131.960, 132.086, 132.544; MS: m/z = 344.91 (M+1).
5-[6-fluoro-2-(thiophen-2-yl)quinolin-4-yl]-1,3,4-thiadiazol-2-amine (5c)
Mp: 149℃ to 153℃; Yield: 68%; IR(KBr: υmax/cm-1): C=C(1640cm-1), 3358(C-NH), 1594(C=N), 3500-3300cm-1(N-H), 1690(N=C), 1460-1006cm-1(C-F); 1H NMR (400 MHz) δ (ppm): 7.229(D,1H, J=4.8Hz), 7.8079(T, 1H, J=6.8Hz), 8.108(D, 1H, J=3.6Hz),8.240(D, 1H, J=3.6Hz), 8.475(S,1H), 8.555(D, 1H, J=5.6Hz), 8.608(S,1H), 8.824(S,2H). 13C NMR (400 MHz) δ (ppm): 118.764, 118.861, 119.902, 120.992, 124.096,128.513, 128.688, 129.048, 129.203, 130.672, 131.422, 131.927, 132.453, 151.755; MS: m/z =328.95 (M+1).
5-[6-nitro-2-(thiophen-2-yl) quinolin-4-yl]-1,3,4-thiadiazol-2-amine (5d)
Mp: 120℃ to 122℃; Yield:62%; IR(KBr: υmax/cm-1): C=C(1640cm-1), 3358(C-NH), 1594(C=N), 3500-3300cm-1(N-H), 1690(N=C), 1600-1400(N=O); 1H NMR (400 MHz) δ (ppm):7.284(D,1H,J=4.8Hz), 7.655(D, 1H, J=7.6Hz),7.914(S, 1H), 8.247(D, 1H, J=8.8Hz), 8.753 (S, 1H), 9.010 (S, 2H) 13C NMR (400 MHz) δ (ppm):129.062, 129.364, 131.650, 139.317, 139.531, 139.735, 139.949, 140.153, 140.367, 140.572; MS: m/z=328.86 (M+1).
5-[6-bromo-2-(thiophen-2-yl) quinolin-4-yl]-1,3,4-thiadiazol-2-amine (5e)
Mp: 98℃ to 100℃; Yield: 70%; IR(KBr: υmax/cm-1): C=C(1640cm-1), 3358(C-NH), 1594(C=N), 3500-3300cm-1(N-H), 1690(N=C), 650-510(C-Br); 1H NMR (400 MHz) δ (ppm): 4.090(S, 2H), 7.225(D,1H, J=6.9Hz), 7.605(D,1H, J=7.6Hz), 7.660(S, 1H), 7.982(T, 1H, J=5.72Hz),8.122(D,1H, J=5.72), 8.226(S, 1H). 13C NMR (400 MHz) δ (ppm): 103.9, 118.1, 121.1, 125.5, 127.6, 127.9, 130.5, 133.5, 140.0, 148.3, 158.3, 161.6, 175.0. MS: m/z=389.78 (M+1) (Figure 1 and Table 1).

Figure 1. Synthesis of 5-[2-(thiophen-2-yl)quinolin-4-yl]-1,3,4-thiadiazol-2-amine
| Codes | Structure of the compound | Molecular formula | Molecular weight | Melting point |
|---|---|---|---|---|
| 5a | 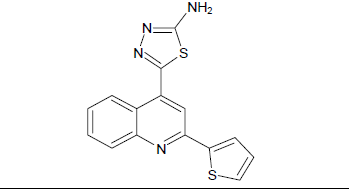
|
C15H10N4S2 | 310.39 | 142-146 |
| 5b | 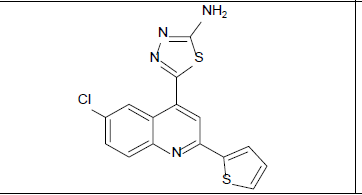
|
C15H9ClN4S2 | 344.84 | 148-152 |
| 5c | 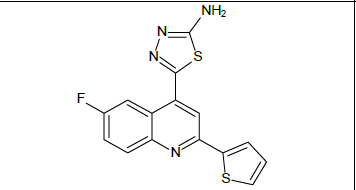
|
C15H9FN4S2 | 328.38 | 149-153 |
| 5d | 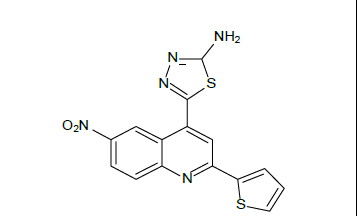
|
C15H9N5S2O2 | 355.39 | 120-122 |
| 5e | 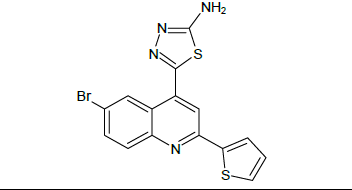
|
C15H9BrN4S2 | 389.29 | 98-100 |
Antioxidant activity
DPPH assay: Oxidized 1, 1-Diphenyl-2-picrylhydrazyl is a stable, purple-coloured free radial. The purple colour, typical for free DPPH radical decays and the changes of absorbance at 520 nm, The spectrometric measurement is followed in the presence of an antioxidant that can provide an electron to DPPH.39,4 mg DPPH dissolved the concentrations of 1 mM stock in 100 ml of methanol stored at 4°C until used in a dark bottle. The DPPH work concentration was 0,14 mM in the test. Diluting methanol 1:1 with deionized water (50 per cent) produced the methanol. Dissolving 2 mg of ascorbic acid and produced up to a total of 10 ml by the deionized water was a standard of ascorbic acid stock I (Conc. 200 mg/mL). A 10 μg/mL concentration range was used for the development of the standard graph of ascorbic acid 2, 4, 6, 8. The DPPH test was conducted using updated method Brand-Williams, adding 140μl of 1 mM DPPH mixed and incubated at 37°C for thirty minutes, in short to a total of 860 tablets per 50 per cent methanol/ascorbic acid/test samples at different concentrations. Read the absorbance by spectrophotometer at 52 nm against 50 percent blank methanol, without the addition of the test samples; a control reaction is performed. Colour correction requires the same concentration of methanol free of the test sample. Acceptable ascorbic acid damping values for antioxidant activity have been calculated. The actual absorption is determined by the absorption of the control and the test sample and the IC50 values (Figure 2) [14].

Figure 2. LEIC50 values for synthesized compounds 5(a-d)GEND
Absorption distribution metabolism excretion prediction
The in silico ADMET study provides an insight into the physicochemical behaviour of the synthesized molecules upon the biological system. The spatial arrangement of the functional group is based on the molecular descriptors and is optimized using QSAR properties. The compounds 5(a-e) are optimized using similar properties. The SAR of 5(a-e) significantly supports us to understand the pharmacokinetics and to establish a relationship between physicochemical properties and predict biological activities based on Absorption, Distribution, Metabolism, Excretion and Toxicity (ADMET). AdmetSAR gives data for the evaluation of active molecules and also for the removal of biologically defective major molecules with unwanted functional groups. The overall analysis of the significant molecules involves surface area, geometry and fingerprint properties, which determine the biological significance of the region in a molecule. Aqueous solubility, intestinal absorption, hepatotoxicity, blood-brain barrier penetration, CACO-2 cell permeability, were the other parameter that helps to understand the metabolic drug mechanism of the molecules synthesized [15].
Drug likeness and bioactive score
The drug-likeness properties of synthesized derivatives 5(a-e), were studied in silico and were calculated by using software’s such as molinspiration cheminformatics. Molinspiration is used to figure out the lipophilicity (log P), TPSA, molecular weight, H-bond acceptors, the number of rotatable bonds, H-bond donors and solubility (log S).
RESULTS AND DISCUSSION
Chemistry
According to the reported method, the compound 2-(thiophen-2-yl)quinoline-4-carboxylic acid and its derivatives 3(a-e) synthesized by using the mixture of 2- acetyl thiophene and Isatin 2(a-e) in ethanol by using catalytic amount of 33% KOH solution. The compound 2-(thiophen-2- yl)quinoline-4-carboxylic acid 3(a-e) and Thio semi carbazide, is taken in catalytic amount of POCl3 acid. The reaction mixture is kept for stirring at temperature 100℃ to 110℃ to obtain 5-[2-(thiophen-2-yl) quinolin-4-yl]-1,3,4-thiadiazol-2-amine and its derivatives 5(a-e) in good yields. The detailed synthetic view is given in scheme. The entitled compounds are purified by column chromatography (using ethyl acetate and n-hexane as eluent). The compounds 5(a-e) was confirmed on the basis of IR, 1H-NMR, 13C-NMR and mass spectral data. All synthesized compounds showed absorption bands ranging from 1640cm-1 for C=C aromatic stretching, 3358 cm-1 for C-NH stretching, 1594 cm-1 for C=N stretching, 3500 cm-1 to 3300 cm-1 for N-H stretching, 1690 cm-1 for N=C stretching, 785 cm-1 to 540 cm-1 for C-Cl stretching, 650 cm-1 to 510 cm-1 for C-Br stretching. 1460 cm-1 to 1006 cm-1for C-F stretching, 1600-1400 for N=O cm-1 stretching of compounds 5(a-e). In 1H-NMR, the presence of singlet peak between δ=4.394-5.854 ppm for –NH2, δ=6.249-6.679 ppm for -N=CH and δ=7.067-9.010 ppm for –C=CH of all synthesized compounds 5(a-e). The triplet Peaks at 7.178-7.982 ppm for S-CH-CH protons of compounds 5(a-e).
Docking
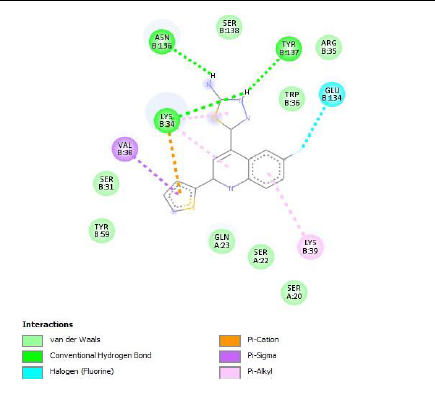
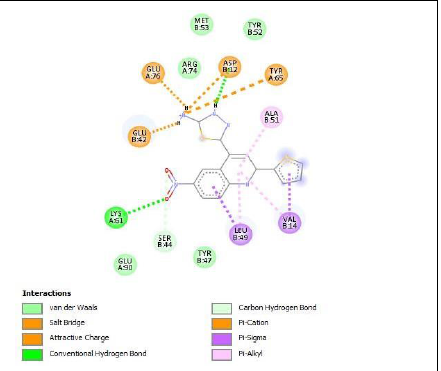
To understand the ligand orientation and interaction capacity of antioxidant targeted ligands. We initially carried out the ligand docking against protein (3VB8) to predict its interaction capacity. The docked ligands vary a binding affinity -7.7 Kcal/Mol to - 6.9 Kcal/Mol (Tables 2 and 3).
| Code | Binding affinity | 3D Interaction | 2D Interaction |
|---|---|---|---|
| 5a | -6.9 | 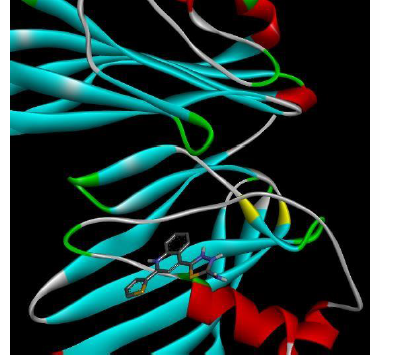
|
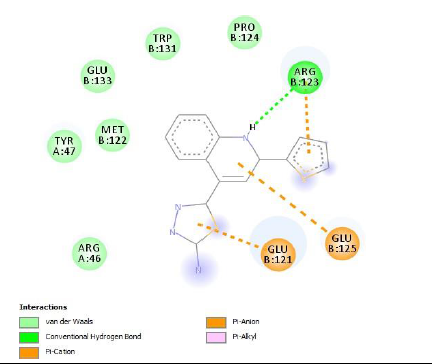
|
| 5b | -7.3 | 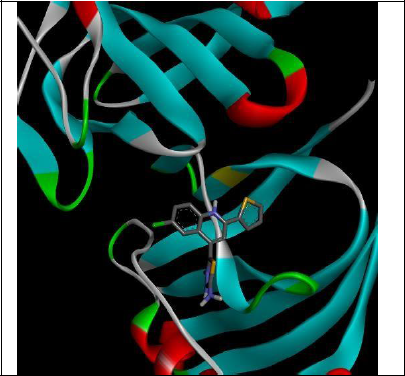
|
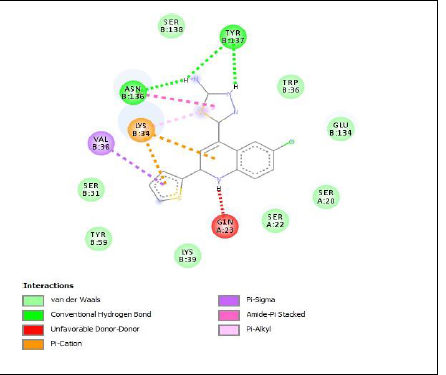
|
| 5c | -7.7 | 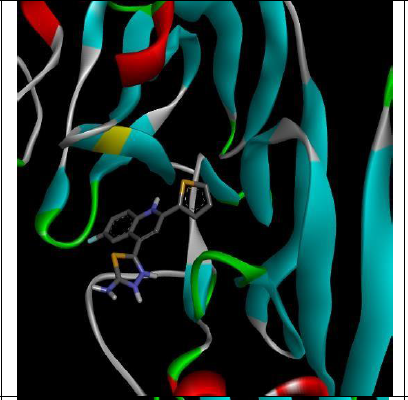
|
|
| 5d | -7.4 | 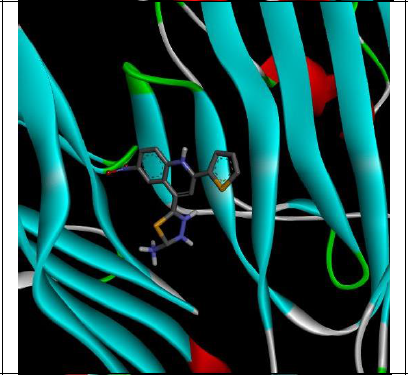
|
|
| 5e | -7.2 | 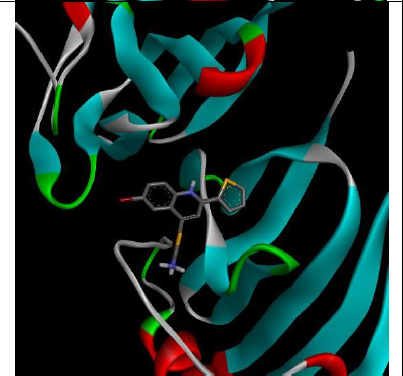
|
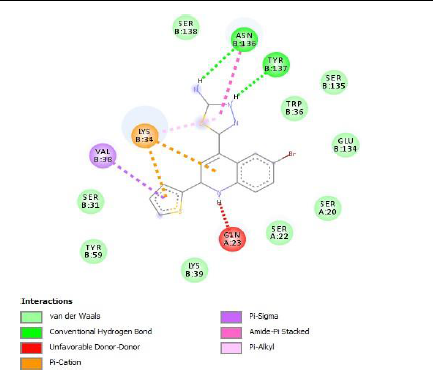
|
| Sl. No. | Samples | DPPH activities (IC50) |
|---|---|---|
| 1 | 5a | 415 µg/ml |
| 2 | 5b | 359 µg/ml |
| 3 | 5c | 375 µg/ml |
| 4 | 5d | 396 µg/ml |
DPPH was used to study the radical scavenging effects of our plant extracts with various solvents with a stable free radical at a feature absorption of 520 nm. The reduce in absorption is taken as a measure of the level of radical scavenging. In the absence of extract, the Radical-Scavenging Activity (RSA) values were expressed as the ratio of the sample absorption decreasing at 520 nm. The results show that 5b showed a 359 μg/ml IC50 value and 5c shows a 375 μg/ml IC50 value. Both samples have been shown to be effective in inhibiting DPPH activity. IC50 values are 415 μg/ml for 5a and 5b and 396 μg/ml.
ADME studies
Pharmacokinetic (PK) ADME properties are one of the primary descriptors in drug discovery for human therapeutic use. These ADME descriptor properties were calculated for the molecules with biological important and are compared with the ranges acquired for standard drugs. The molecule 5- [2-(thiophen-2-yl)quinolin-4-yl]-1,3,4-thiadiazol-2- amine and its derivatives 5(a-e) do possess a considerable degree of hydrogen bond donors and acceptors as recorded through in silico data. The derivatives have been designed to achieve an increase in binding capacity by hydrogen bonding to the receptors. The molecules synthesized accept Lipinski’s rule 5 for oral bioavailability and characters, which enhances their chance to be considered as future drug candidates. The polar surface area on the derivatives was calculated to estimate the ability to permeate cell membranes of different kinds to exhibit their effectiveness. Lipophilicity the ability to transverse through membranes (octanol solubility; water solubility) is measured through log P, is implicated in BBB barrier penetration and permeability prediction in silico. Even the excretion of drugs is dependent on molecular weight and lipophilicity log P. the data generated through the calculation of TPSA are in well within the acceptable range. The overall distribution of compounds in the human body was accessed through by the predicted CACO-2 permeability and BBB coefficient (logBB) and found that the calculated BBB values are in the acceptable range. The values generated for the synthesized derivatives 5(a-e) (Table 4).
| Ligands | logBB | logHIA | Caco | logpGI (substrate) | logPGI (non- inhibitor) | logS | logpapp |
|---|---|---|---|---|---|---|---|
| 5a | 0.9228 | 1 | 0.5619 | 0.7549 | 0.857 | -2.8633 | 1.276 |
| 5b | 0.9248 | 1 | 0.588 | 0.7655 | 0.854 | -3.5561 | 1.4161 |
| 5c | 0.9344 | 1 | 0.5573 | 0.7488 | 0.7362 | -3.3226 | 1.33 |
| 5d | 0.8915 | 0.9962 | 0.5105 | 0.7671 | 0.8695 | -3.1988 | 1.2617 |
| 5e | 0.9164 | 1 | 0.5549 | 0.756 | 0.7751 | -3.4659 | 1.3403 |
| Note: logBB: Coefficient of partition of the blood/brain barrier (1-high penetration, 2-medium penetration and 3- low penetration) | |||||||
| logHIA: Human Intestinal Absorption in nm/s (acceptable range: 0 low, >1 good) | |||||||
| Caco: CACO-2 cell permeability in nm/s (acceptable range; -1 is bad, 1 is good) | |||||||
| logpGI (substrate): P-glycoprotein substrate in nm/s (acceptable range of-5 is poor, 1 is good) | |||||||
| logPGI (non-inhibitor): P-glycoprotein inhibitor (range accepted: 0 to 1) | |||||||
| logs: Aqueous solubility (the amount of concern is 0-2 strongly soluble) | |||||||
| logpapp: Probability of CACO-2 cell permeability in cm/s (1 to 1) | |||||||
Drug likeness and bioactive score
The five rules of Lipinski’s are used predominantly by drug discovery teams in design a drug and their development for their oral bioavailability. The rule insists that, a drug candidate is likely to be orally active, if:
•The drug candidates molecular weight is less than 500
•The partition coefficient (Log P) <5
•H bond donors <5 and
•H bond acceptors <10
•TPSA <160 A0
The molecular properties of 5-[2-(thiophen-2-yl)quinolin-4-yl]-1,3,4-thiadiazol-2-amine and its derivatives 5(a-e) were obtained by molinspiration cheminformatics software. All analogues have not violated any rules of Lipinski and can be expected to be active orally. The lipophilicity is indicated by Log P (partition coefficient) and found that all the compounds were found to be below five indicating clear non-violation of Lipinski’s rule of five.
The molecular weights of 5(a-e) are below 500 and are expected to be transported, diffused and absorbed across the membranes pretty easily than the macromolecules. The synthesized compounds 5(a-e) correspond to the Lipinski rule, which is less than 10 and 5, respectively concerning the number of hydrogen bond acceptors and hydrogen bond donors. TPSA data of the molecules which are correlated with hydrogen bonding and are an indicator for bioavailability orally as the data is in the range of 64.70 Å to 110.52 Å well below 160 Å the limit.
The Molinspiration cheminformatics is the software approached to predict the bioactivity score of the title compounds 5(a-e) for drug targets and is represented.
It is the criteria where a molecule with a bioactive score of more than 0.00 is more likely to show an appreciable degree of bioactivity, while the values between 0.50 to 0.00 show moderate activity and any score less than 0.50 is considered to be inactive. The physiological functions through the obtained data of synthetic molecules are because of the involvement of multiple mechanisms, such as interactions with inhibiting protease, nuclear receptor ligands, GPCR ligands and other enzymes. The data also show that there is substantial interaction between the molecules and the drug targets. Molecules have shown a strong bioactivity score (Tables 5 and 6).
| Compounds | MW | mi LogP | TPSA | No. atom | n-ON | n-OHNH | n-Violation | n-rotb |
|---|---|---|---|---|---|---|---|---|
| 5a | 310.41 | 3.97 | 64.7 | 21 | 4 | 2 | 0 | 2 |
| 5b | 344.85 | 4.62 | 64.7 | 22 | 4 | 2 | 0 | 2 |
| 5c | 328.4 | 3.13 | 64.7 | 22 | 4 | 2 | 0 | 2 |
| 5d | 355.39 | 3.9 | 110.52 | 24 | 7 | 2 | 0 | 3 |
| 5e | 389.3 | 4.75 | 64.7 | 22 | 4 | 2 | 0 | 2 |
| Note: MW: The compounds molecular weight | ||||||||
| mi LogP: Logarithm for the coefficient of partition between water and n-octanol | ||||||||
| TPSA: Polar surface topology | ||||||||
| No: Number of atoms present in compounds | ||||||||
| n-ON: Number of accepters of hydrogen bonds | ||||||||
| n-OHNH: The number of hydrogen bond donors | ||||||||
| n-rotb: Number of bonds which can be rotated | ||||||||
| Compounds | GPCR ligand |
Ion channel modulator | Kinase inhibitor | Nuclear receptor ligand | Protease inhibitor | Enzyme inhibitor |
|---|---|---|---|---|---|---|
| 5a | -0.36 | -0.6 | 0.27 | -0.58 | -0.66 | -0.08 |
| 5b | -0.33 | -0.59 | 0.26 | -0.55 | -0.63 | -0.12 |
| 5c | -0.3 | -0.59 | 0.31 | -0.49 | -0.63 | -0.08 |
| 5d | -0.43 | -0.59 | 0.15 | -0.57 | -0.65 | -0.19 |
| 5e | -0.47 | -0.71 | 0.23 | -0.7 | -0.76 | -0.18 |
Coupled G-Protein Receptors (GPCR): A molecule with a bioactivity score of greater than 0.00 is likely to demonstrate substantial biological activity, while values -0.50 to 0.00 are assumed to be moderately active, and where the score is less than -0.50 it is considered to be inactive.
CONCLUSION
It is the criteria where a molecule with a bioactive score of more than 0.00 is more likely to show an appreciable degree of bioactivity, while the values between 0.50 to 0.00 show moderate activity and any score less than 0.50 is considered to be inactive. The physiological functions through the obtained data of synthetic molecules are because of the involvement of multiple mechanisms, such as interactions with inhibiting protease, nuclear receptor ligands, GPCR ligands and other enzymes. The data also show that there is substantial interaction between the molecules and the drug targets. Molecules have shown a strong bioactivity score.
REFERENCES
- Daniel L, Fabiano SR, Dandara RM, et al., Bioorg Med Chem. 2012, 20: p. 2647.
- Karthik N, Nithiya S, Jayabharathi J, et al., Int J Drug. 2011, 3: p. 123.
- Sharma HL, Sharma KK. Paras Med Publ. 2011, 2: p. 177.
- Yoshikawa T, Naito Y. Jap Med Asscn J. 2002, 45(7): p. 273.
- VL Raman, N Sridevi. J Dnt Med Sci. 2015, 10: p. 109.
- L Langseth. Int Life Sci. 1996, 1: p. 1.
- Sulekha M, Satish Y, Sunita Y, et al., J Chem Phrml Res. 2009, 1(1): p. 102.
- Sies H. Exp Physiol. 1997, 82(2): p. 292.
- Herrera E, Barbas C. J Physio Biochem. 2001, 57(2): p. 45.
- Packer L, Weber SU, Rimbach G. J Nut. 2001, 131(2): p 369S.
- Feixiong C, Weihua L, Yadi Z, et al., J Chem Inf Model. 2012, 52: p. 3099.
- Lin JH, Clin M. Pharmacokine. 2003, 42: p. 59.
- Lipinski CA. Drug Discov Today Technol. 2004, 1: p. 337.
- Pund AA, Shaikh MH, Chandak BG, et al., Polycyc Arom Comp. 2022, 8: p. 1-6.
- Skrzypek A, Matysiak J, Karpińska M, et al., Bioorg Chem. 2021,107: p. 104617.



