Research Article - Der Pharma Chemica ( 2024) Volume 16, Issue 2
Current Trends and Glimpse for All Pharma Global Regulatory Agencies Requirements of Nitrosamine and NDSRIs Impurities in Pharmaceuticals
Rashid Azeez*, Kalyani Bhor and Vinod A BairagiRashid Azeez, Department of Pharmaceutical Quality Assurance, KBHSS Trust Institute of Pharmacy, Bhaygaon, Malegaon Camp, Malegaon Nashik, India, Email: drrashid.azeez@gmail.com
Received: 05-Apr-2024, Manuscript No. Dpc-24-131405; Editor assigned: 10-Apr-2024, Pre QC No. Dpc-24-131405 (PQ); Reviewed: 24-Apr-2024, QC No. Dpc-24-131405; Revised: 25-Apr-2024, Manuscript No. Dpc-24-131405 (R); Published: 16-May-2024, DOI: 10.4172/0975- 413X.16.2.263-273
Abstract
The USFDA and EMEA announced in July 2018 that N-Nitroso Dimethylamine and N-N-NDMA are concentrated on pharmaceutical medicinal products and specifically used in case of SARTANS that are used in the treatment of hypertension and angiotensin II receptor blockers, bringing the seriousness of nitrosamine presence to light. Subsequently, the diabetes medication pioglitazone and the histamine-2 blocker ranitidine were added to the list. Nitrosamines are created when amines, secondary amide carbamates and urea derivatives react with nitrates and nitrogenous agents. Nitrogen is at an oxidation state of +3. Pharmaceutical product degradation, catalysts, solvents, chemical reagents, cross-contamination, the manufacturing process and raw material contamination are some of the causes of nitrosamine presence in pharmaceutical products. Nitrosamine Impurity is detected using technologies such as gas chromatography, mass spectroscopy and light chromatography mass spectroscopy. According to ICH recommendations, N-nitrosamines are classified as a "cohort of concern" because they may be carcinogen and mutagen. The natural world. Class 2A human substances include N-nitroso dimethylamine and N-nitroso diethylamine. The possibility of N-nitrosamine contaminants in pharmaceutical goods poses a problem for the control of medical product quality. N-nitrosamines are regarded as substances of concern since many of the structurally straightforward molecules in this class have strong carcinogenic properties.
Keywords
Regulatory guideline; Risk assessment; NDSRIs; Hypertension; Carcinogen; Mutagen; Nitrosamine impurity; ICH M7; Class 1; Limit
Introduction
Pharmaceutical impurities are unwanted elements that remain in drug product formulations or Active Pharmaceutical Ingredients (APIs). Pharmacological substances may contain impurities that were created during synthesis. Many commonly used excipients have nitrite impurities at ppm levels, which can lead to the development of nitrosamine impurities in drug materials during the manufacture and storage phases. Pharmaceutical companies are now looking for nitrosamine impurities in their goods since these compounds are concerning because they have been linked to human cancer. The primary causes of amine contamination, which can result in secondary, tertiary and quaternary nitrosamines, raw materials sourced from vendors, recycled materials, searing and a loss of method management and control are the main factors that cause nitrosamine to form in drug products. Despite the fact that they can also be found in a variety of foods and liquids, it is seen unsuitable to use them in medications. The following guidelines are recommended for API and pharmaceutical product companies and they address the likely sources of nitrosamine production:
• Perform a risk analysis of their approved or commercialized pharmaceutical products.
• Based on the agency's present understanding, take action to minimize or completely remove nitrosamines from pharmaceuticals.
Nitrosamines were detected in a variety of drug goods, prompting an extensive investigation of these contaminants in the affected pharmaceutical items by the FDA and other international organizations. The FDA has been looking at the nitrosamine contaminants that can be found in several medications. In collaboration with international regulatory partners, the FDA has set globally recognized guidelines for the acceptable daily intake of nitrosamines. Nitrosamine concentrations in drugs under this threshold are acceptable. In cases when the amounts of nitrosamine surpass the daily limit that can be consumed, the FDA recommends that the manufacturer recall the drug. Out of an abundance of caution, some pharmaceutical companies have recalled certain prescriptions, while others have done so because tests showed amounts of nitrosamine exceeding the daily intake limits that are allowed. Based on both rodent carcinogenicity and mutagenicity evidence, ICH M7 (R1) classifies nitrosamine impurities as class 1, which is known to be mutagenic and carcinogenic. Through chromosomal breakage, rearrangements, covalent binding or insertion into the DNA during replication, these nitrosamine contaminants alter the genetic material. Cancer may result from these alterations in genetic material brought on by exposure to extremely low concentrations of nitrosamine contaminants. To protect public safety, it is crucial to detect nitrosamine contaminants in medications at extremely low concentrations [1].
The origins and significant events of nitrosamine impurity profiling
The organic compounds known as nitrosamines or more formally N-nitrosamines, have the chemical formula R2 N=O, where R is frequently an alkyl group. They have a deprotonated amine attached to a Nitroso group (NO+).11 The first description of a class of compounds known as nitrosamines was published in the chemical literature more than a generation ago. Nitrosamines have gained widespread recognition ever since diethyl amine hydrochloride and sodium nitrite were mixed to create Nitrosodiethylamine (NDEA). However, these compounds received little attention from the scientific community until 1954. John Barnes and Peter Magee reported that the substance had hepatotoxic effects when two occurrences of liver cirrhosis among three individuals working in a research facility at an industrial company where NDMA (N-Nitroso Dimethyl Amine) had been introduced as a solvent first occurred that year. A few years later, in 1956, Barnes and Magee also discovered that NDMA was hepato-carcinogenic in rats. Following the discovery that NDMA was carcinogenic, there was a great deal of emphasis focused on the toxicity of Nnitroso compounds. The most common locations for tumor formation in animals are the mouth, throat, stomach, bladder and brain. NDMA and related nitrosamine chemical contamination has been a problem for pharmaceutical companies and the FDA ever since it was found in a small number of valsartan products manufactured in China in July 2018. Later, as extra ARBs (Angiotensin II receptor antagonists), losartan and irbesartan were recalled. The government has been working with manufacturers to prevent the addition of nitrosamines to these products. It was decided that some production processes were flawed. Spartans such as candesartan, irbesartan, losartan and olmesartan are examples of Angiotensin II Receptor Antagonists (ARBs). The existence of NDMA in valsartan and other drugs is being examined. These drugs are used to treat patients with hypertension as well as some cardiac or renal diseases. Nitrosamines became a new cause for concern in the early 1960’s when liver abnormalities were observed in a range of farm animals that had received meals containing herring that had been preserved by the addition of high quantities of sodium nitrite. The dangerous substance in the food could be isolated and identified as NDMA [2].
It was eventually established that the presence of that pollutant was the result of a combination of dimethylamine, an amine found in fish naturally and even sodium nitrite-based nitro sating agent. As a result, researchers began to look into the possibility that human diet may also include nitrosamines. After that, they began to measure the amounts of nitrosamines in human food supplies and the results showed that these contaminants were present in some form in a variety of meals. During manufacturing, the ring structure of these ARBs, tetrazole, may result in the creation of nitrosamine impurities. Nitrosamines can arise during specific processes that take place during the production of ARBs. The FDA had made public the process for comparing ARBs to contamination with nitrosamine in the APIs, such as valsartan, which has been the subject of inquiry and recalls since 2018. Nonetheless, the FDA has consistently overstated the harm that the contamination brings to the patients, citing research that showed little to no increased risk of cancer. In spite of this, ARB issued more recalls up until 2019. Nitrosamine-containing impurities have lately been found in goods that also include ranitidine and pioglitazone. Because ranitidine contains significant levels of NDMA, the FDA is actively monitoring this relatively new pharmaceutical molecule for patient safety. The US regulatory update from October 2, 2019 states that when ranitidine formulations were tested at higher temperatures using a test method that was altered by a third-party laboratory, very high quantities of NDMA were created. In the agency's limited, preliminary testing, ranitidine samples have not yet tested positive for detectable levels of NDMA. When the FDA announced the ranitidine contamination, it said it was still determining how much of a risk to patients [3].
Nitrosamine impurity
A class of chemicals, with the chemical structure of a nitroso group attached to an amine (R1N(-R2)-N=O), is referred to as nitrosamines. A nitrosating reaction between amines (secondary, tertiary or quaternary amines) and nitrous acid (nitrite salts in an acidic environment) can produce the chemicals.
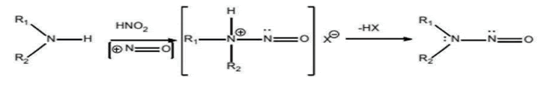
N-Nitrosodiethylamine (NDEA), N-Nitroso-N-Methyl-4-Aminobutanoic Acid (NMBA), N-Nitrosoisopropylethyl Amine (NIPEA), NNitrosodiisopropylamine (NDIPA), Nnitrosodibutylamine (NDBA) and N-Nitrosomethylphenylamine (NMPA) are the seven nitrosamine impurities that the FDA has identified as potentially present in drug products (Figure 1). Five of them the presence of (NDMA, NDEA, NMBA, NIPEA and NMPA) has been found in drug substances and drug products.

Figure 1. Chemical structures of seven potential nitrosamine impurities in APIs and drug products.
The International Agency for Research on Cancer (IARC) has classed some nitrosamine compounds as likely or suspected 17 human carcinogens. Nitrosamine compounds are strong genotoxic agents in a number of animal species. These are known as "cohort of concern" compounds according to the ICH guideline M7(R1) for industry. In order to reduce the possibility of cancer, pharmaceuticals should evaluate and control DNA reactive (mutagenic) contaminants (March 2018) According to the guidelines, any known mutagenic carcinogen, such as nitroso compounds, should be controlled at or below a level where exposure to potentially mutagenic contaminants would not significantly increase the risk of human cancer. The FDA issued interim acceptable limits for these impurities after nitrosamine pollutants were found in ARB’s [4].
Why nitrosamine contaminants are present in APIs?
According to recent data acquired by the FDA, there are multiple common underlying reasons why nitrosamine impurities can exist in APIs:
Condition for formation of nitrosamine impurities: Under acidic reaction circumstances, nitrosamines can form when secondary, tertiary or quaternary amines and nitrite salts are present. Nitrite salts may produce nitrous acid in these circumstances, which can then combine with an amine to produce nitrosamine. If left over azide (a reagent frequently used in tetrazole ring creation or insertion of azide functional group into a molecule)
is quenched with nitrous acid in the presence of precursor amines, there is an increased probability of nitrosamine production. Despite purification procedures, nitrites used as reagents in one stage can persist into following phases and react with amines to produce nitrosamine contaminants.
Thus, carryover into later phases cannot be ruled out anytime nitrite salts are present.
Sources of amines that can form nitrosamine in the secondary, tertiary and quaternary stages: There are several reasons why amines could be used in a manufacturing process. Secondary or tertiary amine functional groups may be present in the starting materials, intermediates or API (or API degradation ants). As reagents or catalysts, tertiary and quaternary amines can also be purposefully added.
Nitrosamines can be created by the reaction of any of these amine types with nitrous acid or other nitro sating chemicals. Another source of secondary amines is amide solvents, which can degrade in specific reaction circumstances. For instance, N,N-dimethylformamide can break down into dimethylamine under high reaction temperatures and long reaction times. Dimethylamine then reacts with nitrous acid to form NDMA (Figure 2). The degradation processes of N-methylpyrrolidone, N,N-dimethylacetamide and N,N-dimethylacetamide also result in secondary amines, which can react with nitrous acid to create nitrosamine contaminants. A mixture of contaminants, including secondary amines, may exist in amide solvents. For instance, N,N-dimethylformamide may include dimethylamine, an impurity that can react with nitrous acid to form NDMA.

Figure 2. Formation of NDMA From N,N-dimethylformamide.
Other amine impurities may be present in tertiary and quaternary amines that are utilized as reagents in the manufacture of APIs. It has been demonstrated that tertiary amines, such triethylamine, include trace amounts of other secondary amines, like dipropylamine and isopropyl ethylamine. Third- and fourth-order amines can exist as contaminants or degradation products resulting from the dealkylation of quaternary amines. For instance, tributyl and dibutyl amine impurities may be present in tetrabutylammonium bromide, a typical phase-transfer catalyst. Each API maker should ascertain the level of amine impurity that could cause nitrosamine contamination of the API, as this varies depending on the method [5].
Contamination in vendor-sourced raw materials: When raw materials and beginning materials obtained from vendors are tainted, nitrosamine contaminants may be introduced. Because of this underlying cause, the agency has noticed the following contaminations:
• Fresh solvents (toluene, methylene chloride and ortho-xylene) have contaminated shipments from vendors (e.g., during transfer between storage vessels) resulting in nitrosamine contamination.
• It is known that certain starting materials, such sodium azide, include sodium nitrite, which can react with amines in an acidic environment to produce nitrosamines.
• Nitrite impurities may be present in nitrate-containing raw materials, such as potassium nitrate. Each API manufacturer should be able to establish the maximum quantity of nitrite impurity that can be tolerated, as this varies depending on the procedure.
• Starting materials or outsourced intermediates may be at risk through cross-contamination if they are manufactured at sites where nitrosamine impurities are produced in other processes.
• Secondary or tertiary amines have been reported as impurities in some raw materials and in fresh solvents such as toluene.
One key to avoiding contamination is being aware of the raw material supply chain. For instance, manufacturers of API’s might not be aware that raw or starting materials they purchased from vendors contain nitrosamine contamination; similarly, a producer whose manufacturing process is not typically prone to nitrosamine formation might not be aware that material purchased from a vendor may have contained impurities introduced during production or transportation [6].
Reagents, catalysts and recovered solvents as potential contamination sources: Recovered materials, like catalysts, reagents and solvents, may contain residual amines (like trimethylamine or diisopropylethylamine) that could lead to nitrosamine impurities. In the event that nitrous acid is utilized to break down any remaining azide during the recovery process, nitrosamines may develop during solvent recovery. Depending on how recovery and further purification are carried out (e.g., aqueous washes or distillation), these nitrosamines may be entrained if their boiling points or solubility properties are comparable to the recovered compounds. This raises the possibility of contamination during material recovery even more. For these reasons, it was discovered that some pharmaceutical items containing APIs produced by specific "low" risk processes were tainted. Because of this underlying cause, the agency has noticed the following contaminations:
• A manufacturing facility may employ many synthetic processes using common solvents to create the same API. The solvents delivered for recovery are at danger if any of those synthetic procedures result in the production of nitrosamines or include precursor amines. Nitrosamine contaminants can be introduced through the careless use of recovered solvents mixed from various manufacturing lines or processes. Even while the synthetic approach is not often prone to nitrosamine production, the API will be compromised if a recovered solvent is contaminated in this way and subsequently utilized to create an API.
• Recovery of raw materials, such as catalysts, reagents and solvents, is frequently delegated to outside contractors. Process outsourcing may be dangerous if the third-party recovery facility only uses standard recovery procedures and does not obtain sufficient specific information about
the materials it is processing.
• If proper equipment cleaning is not done between customers or between different materials or if it is not verified to be capable of eliminating every impurity of concern, raw materials may get polluted. It was alleged that improper cleaning and the usage of shared storage equipment by several customers resulted in the contamination of ortho-xylene and toluene during recovery. If measures are not taken to prevent nitrosamine contamination before materials from other customers are joined for recovery, inadequate and unvalidated cleaning techniques can potentially result in cross contamination. For instance, the combination of catalyst from various customers resulted in contamination of the catalyst tributyltin chloride, which is employed as a source of tri-N-butyl tin azide, at a third-party contractor facility.
A possible source of nitrosamine contamination in the quenching process: When nitrous acid is introduced to the reaction mixture in order to break down remaining azide, this is known as a quenching step that puts the main reaction mixture at risk of forming nitrosamine. This makes it possible for leftover amines in the raw materials used in the manufacturing process to come into direct contact with nitrous acid. If sufficient removal or purification procedures are not in place or if the procedures are not tailored for the removal of particular contaminants of concern, the nitrosamine impurities may be carried to the next phases. Once they are introduced, this can contaminate the entire downstream process. There is a risk if contaminated recovered materials are added to the main reaction mixture, even if the quenching process is carried out outside of it [7].
Lack of process optimization and control: Lack of optimization of the API production process when reaction conditions like temperature, pH or the order in which reagents, intermediates and solvents are added are unsuitable or inadequately controlled is another possible source of nitrosamine impurity development. The FDA has observed cases where the same API's reaction conditions differed significantly between batches and even between different processing equipment within the same facility. The above-mentioned several core reasons of nitrosamine contamination might all happen within the same API process. As a result, it might be required to employ a variety of techniques to find every possible source of contamination. It is doubtful that commonplace testing (such HPLC) for API purity, identification and recognized contaminants will find nitrosamine impurities [8].
Drug products with nitrosamine impurities from sources other than API contamination: Common nitro sating contaminants, such as nitrites, have been found at ppm levels in a variety of excipients. Numerous widely used excipients include nitrite impurities, which can cause nitrosamine impurities to occur in drug products throughout the manufacturing process and during the shelf-life storage period. It is important for the supplier qualification program to consider that nitrite impurities can differ across suppliers and between excipient lots. It is important for drug product makers to be aware that drinkable water can contain nitrite and nitrosamine contaminants. Certain drug products might go through processes of degradation that result in the formation of nitrosamine impurities; this might happen while the drug product is being stored [7].
Nitrosamine impurities' sources
Impurities containing nitrosamines can essentially enter drug substances and drug products by process creation, direct introduction, degradation or cross-contamination. Raw materials, intermediates, solvents, chemicals and reagents are used in the manufacturing of pharmacological compounds. If this impurity forms or is present during these steps, it may be integrated and transmitted to the drug product (Figure 3).

Figure 3. Nitrosamine impurities' sources.
People can be exposed to nitrosamines in a variety of ways, such as through food, drink water, tobacco, personal care products, rubber goods, pesticides and industrial exposure. Regardless of dietary preferences, nitrosamine absorption from food intake has been proven to be the second largest source of nitrosamine exposure, after cigarettes. In addition to being exposed to nitrosamines exogenously, these chemicals can also be created endogenously.
• Nitro sating agents like sodium nitrite, in addition to primary, secondary, tertiary or quaternary ammonium salts, are regarded as precursors
for the production of Nitrosamine impurities. Similarly, nitration of carbamate, amides and N-alkyl amides can result in the formation of nitrosamine impurities. The type, structure and concentration of the nitro sating agent are the primary determinants of the degree of
nitrosamine impurity generation. It is believed that secondary amines are more reactive.
• The process's recovered solvents and catalysts run the risk of forming nitrosamines. Given these nitric acid or sodium nitrite are used to treat
solvents or catalysts in order to eliminate any remaining azide that could cause nitrosamine impurities to form.
• A vendor's contaminated raw material or beginning material may introduce nitrosamine impurities into a drug substance or drug product.
• Nitrosamine impurities may get contaminated by cross-contamination across several manufacturing processes and products on the same production line. The presence of nitrite in the water used in the manufacturing process can taint a process even in cases when nitro sating chemicals are not employed.
• The breakdown of solvents or other materials employed in the manufacture of pharmacological compounds may result in the formation of traces of these contaminants. Similarly, contaminants known as nitrosamines may be introduced into pharmacological compounds as byproducts of the drug production process. Potential NDMA and NDEA impurities can be formed by solvents such Dimethylformamide (DMF), dimethylacetamide or dimethylacetamide.
• Using certain packing materials for the final product could expose it to nitrosamine contaminants. One theory states that amines in printing ink may react with packing material lidding foil containing nitrocellulose to generate nitrosamine impurities, which may then find their way into the medicinal product (Figure 4).

Figure 4. Nitrosamine formation.
Experts speculate that sodium nitrite, which is used to remove leftover sodium azide reagent, may have been the source of the NDMA contaminant in Valsartan. Nitrite ions react with trace amounts of dimethylamine; a by-product of the solvent Dimethylformamide (DMF), to form nitrous acid in an acidic environment demonstrates how contaminants arise during the production of drugs. The following table lists some of the secondary amines and the accompanying Nitrosamine impurities that they created (Table 1).
| Amines | Compounding nitrosamine impurities |
|---|---|
| Dimethylamine | N-Nitrosodimethylamine (NDMA) |
| Diethylamine | N-Nitrosodiethylamine (NDEA) |
| Dipropylamine | N-Nitrosodipropylamine (NDPA) |
| Diisopropylamine | N-Nitrosodiisopropylamine (NDIPA) |
| Dibutyl amine | N-Nitrosodibutylamine (NDBA) |
| Ethyl methylamine | N-Nitrosomethylethylamine (NMEA) |
| 4-(methylamino)butanoic acid | N-Nitroso-N-Methyl-4-Aminobutyric Acid (NMBA) |
Table 1: Amines and corresponding nitrosamine impurities.
There are few cases of sources/pathways published
• Cross-contamination, which occurs on a production line when multiple operations are carried out consecutively.
• Making use of certain packaging supplies. The nitrosamine contamination has been discovered in finished pharmaceutical formulations stored in blister packs with nitrocellulose-containing lidding foil (Figure 5).

Figure 5. The probable origins of Nitrosamines.
Regulations related to nitrosamine
European Union (EU): The European Union (EU) evaluated sartans at risk of harboring nitrosamine impurities (those having a tetrazole ring). Manufacturers were asked to review and adapt their manufacturing procedures to decrease nitrosamine impurities to the extent practically achievable. There will be a two-year transitional period before these policies are put into action. In this transitional phase, items are subject to interim limits, as indicated in Table 1. Products that surpass these limitations for a single contaminant or that contain both NDMA and NDEA in one batch is forbidden in the EU. Testing for nitrosamines is now being added to the drug substance descriptions for the sartan family in the European Pharmacopoeia. The required tests will also be included in the general monograph for APIs, which is presently undergoing revision. These proceedings resulted in the temporary removal of several sartan medications from the EU market. Now that many have returned to the market, the EU has urged customers to keep shopping taking their prescription drugs. The FDA has released a list of ARB medications in a manner akin to how it attempted to locate and recall pharmaceuticals with nitrosamine contents higher than what was deemed appropriate. Both the EMA and the USFDA stressed that there is less risk in continuing the prescriptions with these contaminants than there is in quitting them suddenly and suffering negative side effects, such a stroke. It has been discovered more recently that certain batches of ranitidine and nizatidine products had NDMA impurity levels. Ranitidine drugs are often used to regulate stomach acid production in patients suffering from conditions such as ulcers and heartburn. There are variants available without a prescription and over-the-counter. Regulatory agencies have responded in a variety of ways. A number of specialized national regulatory organizations in Europe, including swiss medic and health Canada, took proactive steps to either ban or restrict the sale of any ranitidine products until batch studies demonstrate that NDMA levels were within allowable bounds. The EMA is now evaluating the available evidence to ascertain if patients on ranitidine are at any risk from NDMA [8].
USFDA: The US Department of Health and Human Services houses the FDA, which is entrusted with "protecting the public health by maintaining the safety, efficacy and assurance of human and veterinary medication products, biological products and medical devices..." The FDA regulates prescription drugs with brand names, generic drugs and Over-The-Counter (OTC) drugs that are not prescribed. When a medication is approved by the FDA, it means that the FDA believes it to be safe and effective. However, problems do occur that compromise the security of authorized products. When problems occur that affect the effectiveness of pharmaceutical products, a drug recall may follow. A drug recall is defined as "a voluntary action taken by the corporation at any time to remove a defective drug product from the market." The FDA or the pharmaceutical business will either declare a recall when a product is thought to be hazardous for patients or when FDA requirements have not been followed.
Among the numerous reasons for medication recalls are incorrect labelling, contamination, the presence of pollutants and a lack of sterility. In the past two years, the FDA has announced 226 medication recalls, according to one count. Considering that "not all recalls are published on FDA.gov or in the mainstream media," there are most likely considerably more drug recalls overall. In June 2018, the FDA received information about valsartan from a single source about the production of pharmaceutical compounds and the presence of an impurity known as N-Nitroso Dimethylamine (NDMA). Further FDA examination found that prescription drugs from different manufacturers of valsartan and other pharmaceuticals also contained N-Nitrosodiethylamine (NDEA), another Nitrosamine contaminant, at insufficient amounts. Since there was no acceptable limit for nitrosamines in the specification, the FDA announced "interim acceptable limits" for these nitrosamine impurities in ARB drugs as a crucial step. It was suggested to eliminate medicine ingredients and drug [9].
Other regulatory agencies
Guidelines on the presence of nitrosamine impurities in "sartan" blood pressure medications were released by the Therapeutic Goods Administration (TGA) of Australia.
Limit of nitrosamine detected by various agencies: It recommends that when establishing nitrosamine impurity limits for APIs and pharmaceutical products, manufacturers should make use of these AI technologies. These limits are present in following Table 2:
| Nitrosamine | AL Limit (ng/day) |
|---|---|
| NDMA (N-Nitroso Dimethylamine) | 96 |
| 8NDEA (N-Nitrosodiethylamine) | 26.5 |
| NMBA (Nitroso Methyl-N-Butylamine) | 96 |
| NMPA (N-Nitrosomethylphenylamine) | 34.3 |
| NIPEA (N-Nitrosoisopropylethylamine) | 26.5 |
| NDIPA (N-Nitrosodiisopropylamine) | 26.5 |
| NDBA (N-Nitrosodibutylamine) | 26.5 |
Table 2: Limit of nitrosamine detected by various agencies.
ANVISA (Brazil):
• Technical and scientific aspects that the FDA should take into account when assessing the risk of NDSRIs in creating best practices for carrying out NDSRI testing in order to assist the establishment of AI limitations (e.g., Ames test, improved Ames test, follow-up in vitro mutagenicity, in vivo transgenic gene mutation test). Additional tests (and techniques) suggested evaluating NDSRIs' mutagenic potential.
The value of "short-term" carcinogenicity testing (such as a six-month transgenic mouse model) in assessing the danger of NDSRIs and the benefits and drawbacks of doing so. Research on DNA biomarkers, reactive intermediate identification, in vitro/in vivo metabolism and other topics that could provide the FDA with more information regarding the risks connected to NDSRIs.
• If the suggested time frame for drug testing confirmation should be extended products and the filing of necessary modifications to drug applications (between October 1, 2023 and June 1, 2024) would facilitate applicant cooperation and the extra evaluation of NDSRIs.
• How the FDA can assist producers in completing certain types of confirmatory testing limits on Acceptable Intake (AI)
• How the FDA can support cooperative efforts to provide trustworthy compound-specific information on NDSRIs, lessening the requirement
for extra (and possibly redundant) testing?
• The challenges that the industry has faced in collaborating to share data for evaluating the safety of NDSRIs, especially in light of the goal of
minimizing redundant testing and incorporating the 3R principles; additionally, the agency can assist stakeholders in overcoming these challenges.
• The inability of suppliers or manufacturers to meet advised AI limitations has resulted in the suspension of medication product distribution or manufacturing.
Materials and Methods
A realistic and evidence-based approach to determining tolerable doses for drug product nitrosamine impurities The TD50 calculator and N-nitrosamine carcinogenicity explorer. A single internal database, known as the Pfizer N-nitroso carcinogenicity explorer, was created by combining nitrosamines whose carcinogenicity statistics were summarized in the Lhasa Vitric database and the Lhasa Carcinogenicity Database (LCDB). The Lhasa carcinogenicity database (lhasalimited.org) is a publicly accessible online structure-searchable database containing carcinogenicity data. With over 1700 chemicals included, the database includes all of the information from the original Carcinogenic Potency Database (CPDB), created by Gold et al., as well as further information selected by Lhasa. The Pfizer N-nitroso carcinogenicity explorer database had 970 studies from 248 N-nitrosamines, 1435 treatment groups and 2612 end points (mg/kg/day) that were specific to tissues and tumors. The database was hosted in a MySQL relational database [10].
Structural organization of complex N-nitrosamine: A large number of drug product N-nitrosamines required estimations of daily AIs to determine as part of the portfolio review of Pfizer which ones need examination through analysis. These structurally complicated N-nitrosamines could be theoretically produced via reaction between a nitro sating agent and secondary amine precursors contained in the API itself or as an impurity in the API and/or drug product degradant. We divided the structurally complex N-nitrosamines into 13 structural groups are as follows (Figure 6).

Figure 6. Typical structural elements found in complex nitrosamine impurities.
Since it was labor-intensive to estimate individual AIs for a large number of impurities using a read-across approach. Based on the structural similarities in the immediate area of the N-nitrosamine substituent and the previously established structure-activity connections for the carcinogenic potencies of N-nitrosamines with low molecular weight. Afterwards, independent visual inspections of the compounds in the Pfizer N-nitroso carcinogenicity explorer were used to select read-across substrates for each structural group. These compounds were chosen because they were structurally similar to members of the different structural groups in terms of their electronic and steric environments. The read-across methodology was then used to determine the daily AIs for each of the 13 structural groupings. For every group, a single worst-case AI was determined based on structural similarities and this AI could be used for any related complex N-nitrosamine that was allocated to the group.
Results and Discussion
Assessment of the carcinogenic potency information The main resource for determining carcinogenic potency information (TD50 values) was LCDB. The initial TD50 values within the LCDB were determined using. Use is used of Gold et al., (herein referred to as Gold TD50). Furthermore, Lhasa Limited independently computed TD50 values (henceforth referred to as Lhasa TD50) using Lhasa's stated methodology and the identical data utilized for the Gold TD50. For carcinogenic Nnitrosamines of interest, the LCDB was searched for accessible TD50 values. The most reliable study that was found met the requirements mentioned in ICH M7(R1). In a nutshell, the optimal research for AI derivation included three dose groups with a minimum of fifty animals in each group, treatment for the animals' lifetime and dosing at least five days a week; However, research that did not satisfy every one of these requirements were still considered for AI derivation in our assessment, especially in cases where there was a strong reaction of the tumor. Since the Lhasa database is up to date and TD50 value computations are clear, Lhasa values were used where both Gold and Lhasa TD50 values were available for a certain N-nitrosamine. Furthermore, since Lhasa involves the derivation of a TD50 from many treatment groups, studies containing TD50 values are often more robust than those containing only Gold TD50 values. Tumor incidence statistics were gathered from literature references in cases where a chemical was not recorded in the LCDB and a TD50 was computed using R code that was modified from Lhasa. To put it briefly, the influence of dose on the chance of tumor occurrence was captured by a linear model that was optimized utilizing the Broyden-Fletcher-Goldfarb- Shannon algorithm within the optimizer function. This model was based on binomial likelihood and eventually estimated the TD50. If more than one study was published, the most robust study's TD50 value representing the carcinogenic potency of each analyzed nitrosamine was taken (in a cautious manner) together with the most sensitive species, sex and target organ from that study. There was no use of the harmonic mean TD50 value [11].
The analytical technique for impurities with nitrosamines
Because there are extremely little amounts of impurities in the complex matrices, developing analytical techniques to identify nitrosamine impurities is a difficult task. To ensure that the created techniques meet GMP requirements, they must also be validated. The FDA has released multiple strategies to address NDMA and NDEA in various "sartans." The EMA has stated that further nitrosamines will be included in the expansion of measures. Several regulatory bodies, including those in Singapore, Switzerland and Canada, have released analytical techniques and implemented their own policies. Nitrosamines in drug substances and drug products are typically tested using chromatographic techniques like Gas Chromatography (GC) or reversed-phase Liquid Chromatography (LC) in conjunction with different detectors like Mass Spectrometry (MS), Ultraviolet Spectrophotometry (UV) or nitrogen chemiluminescence. These published testing protocols provide as a foundation for the creation and verification of analytical procedures suitable for different drug substances and drug products. According to the FDA, whether the results are used in a regulatory submission or to support an obligatory quality assessment of the drug product or API, the user must validate these methodologies.
Gas chromatography: Mass Spectrometry and Gas Chromatography (GC-MS) is the method most commonly used for the identification of smaller molecular weight nitrosamines. Because of its great selectivity and low detection limit, GC-MS, GC–MS/MS or GC-HS-MS technology is used in most current studies. The four nitrosamine impurities in Valsartan drug substance and drug products are N-Nitroso Dimethylamine (NDMA), NNitrosodiethylamine (NDEA), N-Nitrosodiisopropylamine (NDIPA) and N-Nitrosoethylisopropylamine (NEIPA). The FDA has developed and validated a combined GC-HS-MS method for the simultaneous evaluation of these impurities. This technique surpasses the anticipated requirements of the control limits and complies with all current rules with sensitivity and reproducibility. A thermometer with good selectivity for nitrosamines is the GCTEA (thermal energy analyzer). Because of their enormous molecular weight and relative labile nature, it is impossible to ascertain. Liquid chromatography: The LC technique provides a quicker substitute for the conventional GC-MS techniques. In order to acquire adequate selectivity for the identification of GC detectable and GC undetectable molecules as well as thermally stable and unstable Nitrosamines, highresolution accurate mass spectrometry is used. The FDA has noted that because heating the sample produces NDMA, the GC-MS method for testing ARBs of nitrosamine impurities is not appropriate for testing ranitidine. Subsequent to ICH Q2 (R1), the FDA created an LC-HRMS method to evaluate the amounts of NDMA in ranitidine drug substance and drug product. The method's range is 0.033 ppm-3.33 ppm, its Limit of Quantitation (LOQ) is 0.033 and its Limit of Detection (LOD) is 0.011 ppm. Furthermore, a number of techniques utilizing liquid chromatography-mass spectrometry or LC-MS/MS have been documented in the literature of science. Still, very few researches have been published for NDMA analysis utilizing traditional High-Performance Liquid Chromatography (HPLC), particularly in the pharmaceutical industry. It is ideal if NDMA contaminant is found alongside drug constituents in a single HPLC examination. HPLC is the most widely used technique for quality control of APIs and products in routine analysis. Therefore, it's critical to create a quick and easy HPLC testing method for NDMA in medications [12].
Recommendations to API manufacturers
The great majority of APIs are not anticipated to have nitrosamines during production, however all producers of chemically synthesized APIs should take the necessary precautions to reduce the possibility of nitrosamine contamination for APIs that may contain nitrosamine impurities. To find any chance of nitrosamine impurities, API manufacturers had to examine their manufacturing procedures and do risk analyses. In the event that nitrosamine impurity risk is detected, batches should undergo confirmatory testing with sensitive, suitably validated techniques. There is no need to take additional action if the risk evaluation finds no evidence of nitrosamine contaminants. The primary cause of any found nitrosamine impurity should be looked into by API makers. They ought to alter the manufacturing procedure [13].
Reducing nitrosamine impurity presence in API’s: The FDA advises API producers to do the following: Optimize the production process design for APIs during Route of Synthesis (ROS) development in order to reduce or avoid the generation of nitrosamine contaminants. For industry guidance on good manufacturing practices for APIs, manufacturers should consult ICH M7(R1), as well as industry Q7 good manufacturing practice guidance for active pharmaceutical ingredients (September 2016) and Q11 development and manufacture of drug substances (November 2012). When developing a process, the following elements need to be considered:
• Preventing reaction circumstances that could result in nitrosamines whenever feasible; in the event that this is not feasible, proving that the process is sufficiently regulated and capable of reliably lowering nitrosamine contaminants through suitable and strong study on fate and purge.
• If ROS circumstances have the potential to produce nitrosamines, using bases other than secondary, tertiary or quaternary amines (where possible).
• Exerting caution when using amide solvents (such as N,N-dimethylformamide, N,N-dimethylacetamide and N-methyl pyrrolidone) to combat ROS. Using other quenching agents in place of nitrites for azide breakdown procedures.
• Efficient and reliable management of reaction sequences, procedures and reaction conditions (pH, temperature and reaction duration).
• Creating a production procedure that makes it easier for nitrosamine impurities to be removed in later processing stages.
• In order to lower the possibility of nitrosamine production, API makers can think about eliminating quenching stages from the primary reaction mixture (for example, by utilizing nitrous acid to break down remaining azide). The mother liquor can be separated from the organic phase by the API or an intermediate created by a reaction with an azide salt. After the organic phase and aqueous waste phase have been separated, nitrous acid should be used to quench the waste phase without coming into touch with the API, its intermediate or recovery-related solvents.
• Manufacturers of APIs should examine and keep an eye out for any raw materials, beginning materials or intermediates that could be at risk in their supply chains. API producers should keep records that include the supplier's name and that of the raw material manufacturer, functions of the actual producers of these products, as well as any distributors and repackers who work with the components prior to API production. To avoid nitrosamine contamination, API makers should, when applicable, implement controls and consider extra requirements for materials that are considered to be at-risk.
• Recovered materials, such as solvents, reagents and catalysts, should only be utilized in the same step of the same process or in an earlier phase if there is adequate purification, to prevent cross-contamination during the manufacturing process.
• Where it was gathered. Before being used again, the recovered materials must adhere to the necessary criteria. The API maker should examine the contractors' validation of cleaning methods if material recovery is delegated to outside contractors. In order to ensure that cross contamination with nitrosamine or nitrosamine precursors can be avoided, API producers should adhere to the guidelines in ICH Q7. Additionally, API producers ought to confirm with their suppliers if the resources they buy and utilize in their operations are recovered.
• Manufacturers of APIs need to be aware that low concentrations of nitrite and even nitrosamines from environmental contamination may be present in potable water used in API manufacturing. Nitrites in processing water have the potential to contaminate API with nitrosamine produce. Thus, API makers should monitor the levels of nitrite and nitrosamine in water and utilize water that has been treated to remove undesirable impurities in order to prevent APIs from having nitrosamine impurities at unacceptable levels.
Regulation of nitrosamine contamination in APIs: The API producer must devise a plan to guarantee that the nitrosamine level stays within the AI limit if a nitrosamine impurity is found to be present above the LOQ. To make sure that, manufacturers should create a suitable control strategy that includes specification restrictions. The API's nitrosamine level consistently stays substantially below the AI limit. Testing of each batch on release should be carried out for APIs with an impurity discovered above the LOQ or at-risk APIs, given the current concerns surrounding nitrosamine contaminants and their presence in pharmaceuticals. Before being implemented, alternative strategies (such as an upstream test of an intermediate) should be presented to the FDA as a supplement and backed by proof of sufficient statistical control and process understanding. The API maker should not release any batch of API for distribution if it is discovered to contain levels of nitrosamine impurities higher than the advised AI unless the FDA has already agreed that the API is required to mitigate or avoid a drug shortage.
Recommendations to drug product manufacturers
To find out whether their products have the potential to include nitrosamine contaminants, drug product makers should perform risk assessments. In order to identify the API ROS or other process conditions of the API, cooperation with the API manufacturer should be part of the risk assessment process. APIs that increase the possibility of nitrosamine contaminants in the medication product. Evaluation of any mechanism (including degradation) that could introduce nitrosamines during the manufacture or storage of medicinal products should be part of the risk assessment. There is no need to take additional action if the risk evaluation finds no evidence of nitrosamine contaminants. If nitrosamine risk in a drug product is found, batches should be subjected to confirmatory testing with sensitive and well validated techniques. In order to minimize or reduce nitrosamine impurities, manufacturers should look into the underlying source of any found nitrosamine impurities and adjust the manufacturing process.
Management of nitrosamine impurities in pharmaceutical items
When developing a control strategy, drug product makers should consider the possibility of nitrites arising from the use of at-risk APIs in manufacturing processes. It is imperative that they assess the possibility of nitrosamine formation in a therapeutic product during its whole shelf life. Manufacturers should remove the source of contamination if an external source that may be prevented introduces nitrosamines into the drug product. The manufacturer must devise a plan to guarantee that the nitrosamine level stays within the AI limit if a nitrosamine impurity is found above the LOQ. The discovered nitrosamine should have specified limitations as part of the control plan. When the entry of nitrosamine is inevitable because of the structure of the API, the ROS in the API or the pharmaceutical product's or API's manufacturing procedure. Given the current lack of knowledge regarding nitrosamine contaminants and their prevalence in medications, testing ought to be done on each batch before to release. Before being implemented, alternative methods should be submitted to the FDA as a supplement and backed by proof of sufficient statistical control and process understanding. Drug product producers should notify the FDA so that the Agency can decide what regulatory action to take for the individual drug goods if batches of their products have unacceptable levels of nitrosamine contaminants and are already in circulation. The producer of the drug product should not distribute any batch of drugs discovered to have nitrosamine impurity levels at or above the advised AI.
Review of the manufacturing process
The Marketing authorization holders can use templates provided by the European Medicines Agency (EMA) to submit the findings of product testing for nitrosamine contamination. The manufacturer mandates the following actions to regulate the evaluation of nitrosamine in human pharmaceutical products. Risk evaluation: As per ICH Q9 and ICH M7 recommendations, the marketing authorization holder, drug substance and drug product maker shall conduct a risk evaluation of nitrosamine within six months (Notice publication 26 September, 2019). Prioritizing the assessment of risks means focusing on the ones with the highest chance of contamination first. The findings of the evaluation must be communicated to the authorities. In the event that a possible contamination risk is identified, the holder of the marketing authorization should go on to step 2, as shown below. Confirmatory testing: The risk assessment, confirmatory testing ought to get underway right away. As soon as feasible, a high-risk product must be analytically evaluated for nitrosamine impurities using procedures that have been established and are suitably sensitive. Similarly, unless there is a valid reason to do otherwise, confirmatory testing of every drug product in question must be completed no later than three years after the notification is published. It is imperative to promptly notify the appropriate authorities of any detection of nitrosamine, regardless of the quantity found. Modifications to the marketing permission: Modifications to the marketing authorization, such as adjustments to the manufacturing procedure for a medicinal substance or modifications to the specifications of a drug product must be implemented promptly. It is necessary to notify the appropriate authorities right once if there is a risk to the public's health. All steps need to be finished in a prioritized way within three years.
Preventing contamination with nitrosamine
It is believed that all amines and nitro sating agents are precursors to the creation of nitrosamine impurities in the drug's active ingredient or byproducts. Therefore, taking the following safety measures could reduce these contaminants in pharmaceuticals for humans.
• When nitrites or other nitro sating agents react with quaternary ammonium salts or secondary or tertiary amines during one or more steps of the drug-substance production process, nitrosamine impurities are created. Therefore, the creation of nitrosamine impurities can be avoided by not
using these reagents.
• The solvent removes a lot of the contaminants in nitrosamine. There's a potential that these contaminants will reappear during the medication production process if these solvents are recovered and employed again. Consequently, it is best to avoid using recovered solvent in the production process.
• Nitrosamine impurities may also originate from tainted intermediate, reagents and raw materials used in the production of pharmaceuticals. Nitrosamine impurities may arise as a result of the intermediate and raw material degradation products when stored in an environment with nitrite traces. As a result, it is important to store these items correctly and check for nitrosamine contaminants.
• Because of past products, nitrosamine contaminants may cross-contaminate equipment used to manufacture medicinal compounds. It is
important to thoroughly clean and inspect equipment for contamination.
• The maker of the drug substances ought to test and examine the presence of nitrosamine impurities at different intermediate stages and if they are, they ought to be regulated at the appropriate limit.
• The producer ought to alter the procedure to eliminate contaminants such as amines, nitrites and nitrosamine at different phases. The implementation of control measures is necessary to identify and manage nitrosamine contaminants in intermediate or drug compounds.
Pharmaceutical items recalled due to nitrosamine contaminants
Recalls of pharmaceutical products have increased in the last few years. These days, this is a widespread issue and producers must be prepared for any emergencies that may result from ineffective product recalls. Patients and regulatory bodies are extremely concerned about the presence of nitrosamine contaminants in prescription drugs. In the last two years, more than 1400 product batches have been taken off the market or recalled because the amount of nitrosamines they contained exceeded the daily allowable limit. As a result, a number of pharmaceutical drugs including the APIs valsartan, irbesartan, losartan, metformin, ranitidine and nizatidine were recalled or taken off the market. Three distinct kinds of nitrosamine contaminants were discovered to be present in these medication goods. The FDA refrained from recalling the life-saving medications rifampin and rifapentine in order to prevent a shortage on the market. The MNP and CPNP origins of these medications are still being investigated. As per the FDA, the acceptable daily intake range for most individuals to consume nitrosamine is between 26.5 ng and 96 ng. Losartan is the medicine that has been linked to the greatest number of recalls (almost 24%), with 324 batches containing this prescription being removed from distribution. After a thorough analysis of the FDA data, it is discovered that about 81% of recalls involve pharmaceutical products that contain "sartans". Apart from sartans, approximately 42% of the total consisted of combination medicines that combined sartans with other APIs.
EMA's request to investigate the potential for the presence of nitrosamines
The findings of the sartans review indicated that nitrosamines may or may not be present in APIs for other medications, contingent on the manufacturing processes used for the drug product and the API. Consequently, all MAHs for pharmaceuticals containing chemically synthesized active components were explicitly required by EMA in September 2019 to evaluate the risk of N-nitrosamines in their products and put the appropriate risk mitigation measures in place. In the call for review, the following steps ought to be completed:
Step 1: Conduct a risk assessment to determine which active components and pharmaceutical products are most vulnerable to N-nitrosamine formation or (cross-) contamination. Step 2: Items that were demonstrated to be sensitive to nitrosamine generation or contamination should undergo confirmatory testing. Step 3: If the existence of nitrosamine(s) is verified, MAHs should update the dossier as needed.
Determination of global risk
In order to classify the global risk, different impact factors (ratios) were assigned to each of the potential causes listed, based on their expected importance in the global risk. As previously mentioned, this technique was applied to the drug ranitidine genericis 150/300 mg, which was developed during the testing of the drug losartan generics 50 mg /100 mg. Ranitidine, has recently been banned by the EU due to the presence of tiny levels of NDMA. Losartan genericis 50 mg/100 mg was chosen for this study because, when it was first started, it was one of the few medications for which genericis pharmaceutical; S.A. already had a substantial quantity of information about the production process for APIs. Furthermore, this drug is used to treat hypertension, one of the most common chronic illnesses; hence it was believed that the existence of nitrosamine contaminants in this drug would have a detrimental effect on the general public's health. Since ranitidine generis 150/300 mg was one of the goods for which a larger amount of information was already available and because it was one of the newest products with nitrosamine impurity presence at the time, it was chosen.
Conclusion
Extremely carcinogenic and mutagenic nitrosamine contaminants must be kept to a minimum in drug ingredients and drug products. To reduce the amount of nitrosamine impurities in therapeutic substances, potential sources including raw materials, reagents, catalysts, solvents and crosscontamination should be found. Several public alerts have been released by medicine regulatory bodies like the FDA, EMA, TGA and Health Canada to assist manufacturers in controlling and limiting these contaminants to levels of tolerable intake. The most recent identification of nitrosamine contaminants in a number of widely accessible drugs has raised awareness of the mutagenic and carcinogenic potential. Owing to the solvent, catalyst and raw materials used in the manufacturing process, these contaminants formed on the medication. The numerous regulatory bodies have issued directives to modify the control methods of the production process in order to manage various nitrosamine impurities and prevent them. Analytical procedures that have been validated are used to discover impurities in manufactured biological and pharmaceutical goods. The total amount of nitrosamine impurities should not exceed 26.5 ng/day (acceptable intake of nitrosamine) based on the maximum daily dose.
References
- Nilsson R. Regul Toxicol Pharmacol. 2011; 60(2): p. 268-280.
[Crossref] [Google Scholar] [PubMed]
- Lijinsky W, Andrews AW. Mutat Res Fundam. 1983; 111(2): p. 135-144.
[Crossref] [Google Scholar] [PubMed]
- Yang CS, Yoo JS, Ishizaki H, et al. Drug Metab Rev. 1990; 22(2-3): p. 147-159.
[Crossref] [Google Scholar] [PubMed]
- Barnes JM, Magee PN. Br J Sports Med. 1954; 11(3): p. 167.
[Crossref] [Google Scholar] [PubMed]
- MAGEE P, BARNES. J Br J Cancer. 1956; 10(1): p. 114-122.
[Crossref] [Google Scholar] [PubMed]
- Crosby NT, Sawyer R. Adv Food Nutr Res. 1976; 22: p. 1-71.
[Crossref] [Google Scholar] [PubMed]
- Iram M. Indian J Pharm Sci. 2020; 13(1): p. 89-91.
- Rao GS. Pharma Int. 1980; 1: p. 187-190.
- Beard JC, Swager TM. J Org Chem. 2021; 86(3): p. 2037-2057.
[Crossref] [Google Scholar] [PubMed]
- Berrido AM, Byrd JB. Curr Hypertens Rep. 2020; 22: p. 1-8.
[Crossref] [Google Scholar] [PubMed]
- Gold LS, Slone TH, Manley NB, et al. Environ Health Perspect. 1991; 96: p. 11-15.
[Crossref] [Google Scholar] [PubMed]
- Thresher A, Gosling JP, Williams R. Toxicol Res. 2019; 8(5): p. 696-703.
[Crossref] [Google Scholar] [PubMed]
- Parr MK, Joseph JF. J Pharm Biomed Anal. 2019; 164: p. 536-549.
[Crossref] [Google Scholar] [PubMed]



