Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 9
Catalyst Free Synthesis of N-(1,3-Dioxoisoindolin-4-Yl)Acetamide Derivatives using Water and their Biological Evaluation
Rajesh Kumar Rapolu1,2, Ravi Kumar Sadineni1,2, Raja Sekhar Bhupathi1, VVNKV Prasada Raju1, Murthy Chavali2, N Srinivasu2, Varimadugu Aruna3, Chaitanya Mulakayala4 and Naveen Mulakayala5*
1Granules India Limited-R and D Center, Plot No.56, Road No.5, ALEAP Industrial Area, Pragathinagar, Hyderabad 5000072, India
2Division of Chemistry, Department of Sciences and Humanities, Vignan's Foundation for Science, Technology and Research University (VFSTRU; Vignan University), Vadlamudi, Guntur 522 213 Andhra Pradesh, India
3Department of Biotechnology, Chaitanya Bharathi Institute of Technology, Gandipet, Hyderabad-500075, India
4Department of Biosciences, Sri SatyasaiInstitute of Higher Learning, Anantapuram-515001, India
5SVAK Lifesciences, ALEAP industrial area, Pragathi Nagar, Hyderabad 500090, India
- *Corresponding Author:
- Naveen Mulakayala
SVAK Lifesciences
ALEAP industrial area
Pragathi Nagar, Hyderabad 500090, India
Abstract
Green and eco-friendly synthesis of N-(1,3-dioxo-2-phenylisoindolin-4-yl)acetamide derivatives were developed by reacting N-(1,3-dioxo-1,3-dihydroisobenzofuran-4-yl)acetamide with different aliphatic aromatic and heterocyclic amines under catalyst free conditions using water as a eco-friendly solvent. These reactions involve simple work-up with high yields. All the compounds were evaluated activity against TNF-α in vitro. Compound 6d showed potent activity against TNF-α with 78% inhibition at 10 μM concentration. Therefore, these compounds found to be valuable leads against TNF-α.
Keywords
N-(1,3-dioxo-2-phenylisoindolin-4-yl)acetamide, Synthesis, Water, TNF-α, in vitro.
Introduction
Cancer is the one of the challenging diseases for organic and Medicinal chemist to make curable medicines against it. It is the second leading cause of death behind cardiovascular disorders in the top ten leading diseases which cause death in the world. In most of the literature pro-inflammatory cytokine, tumour necrosis factor (TNF) expression were screened against Thalidomide derivatives [1,2]. TNF is a main regulator of the inflammatory cascade controlling several pathways, the important ones are anti-angiogenic, anti-inflammatory and immuno-modulatory. Phthalimides are having several pharmacological properties, such as anti-inflammatory [3], antimycobacterial [4], analgesic [5] and anticonvulsant [6]. Apart from this, phthalimide derivatives acts as immunomodulators [7], angiogenesis inhibitors [8], prostate cancer [9], and against human multiple myeloma [10].
On the other hand, it is very important to develop an eco-friendly method for the synthesis of organic molecules which are biologically important to overcome the chemical pollution and resource depletion. Solvents are one of the key materials in most of the organic reactions. To control the environmental pollution organic solvents are replaced with eco-friendly solvents [11]. Based on the green chemistry principles, a green solvent should meet numerous criteria such as non-volatility, non-flammability, low toxicity and widespread availability among others [12]. According to the literature water [13], acetic acid, glycerol [14], polyethylene glycol [15], ionic liquids [16] are considered as green solvents. Among all the ecofriendly solvents, water considered as best eco-friendly solvent.
As part of our ongoing studies [17-26] we wish to explore the synthesis of N-(1,3-dioxo-1,3-dihydroisobenzofuran-4-yl)acetamides with several aromatic and aliphatic amines using water as a green solvent. Further the synthesized compounds were tested for their biological activity against TNF-α.
Materials and Methods
Experimental section
Melting points are uncorrected and were determined in open capillary tubes in sulphuric acid bath. TLC were run on silica gel – G pre coated plates and visualization was done using iodine or UV light. IR spectra was recorded using Perkin–Elmer 1000 instrument in KBr pellets. 1H-NMR spectra was recorded in DMSO – d6 using TMS as internal standard with 400 MHZ Bruker instrument. Mass spectra was recorded on Agilent-LCMS instrument under CI conditions and given by Q+1 values only.
General procedure for preparation of compound 3 from 1 and 2
To a stirred solution of 3-nitropthalic acid (50.0 g, 0.2368 mol), in 3N NaOH solution was stirred at 70°C-75°C for 15 min, and charged FeCl3.6H2O (3.84 g, 0.014 mol), activated Charcoal (5.0 g) to the reaction mixture, then Hydrazine monohydrate (30.31 ml) was added slowly for 30 minutes and maintained for 1 h. After monitoring the TLC, the reaction mixture was filtered at hot condition through hyflow bed to collect the clear filtrate. The filtrate was cooled to 0°C-5°C and maintained for 1 h. Solid compound was observed and filtered to yield compound 3 as a yellow solid.
General procedure for preparation of 4 from 3
To a stirred solution of 3-amino pthalic acid (40.0 g, 0.0714 mol), in acetic anhydride (220 ml) was stirred at 135°C-140°C for 1 h. After completion of the starting material (reaction was monitor by TLC), the reaction was cooled to RT for 30 min and cooled to 0-5°C. Solid compound formation was observed and filtered. The solid compound was made slurry with MTBE (50 ml) and filtered to get compound 4 as a yellow solid.
General procedure for preparation of 6 from 4 and 5
To a stirred solution of 4 (600 mg, 1.0 eq) and amino compound 5 (1.0 eq) in water (5.0 ml) was stirred at reflux for 1 h. After completion of the starting material (reaction was monitor by TLC) the reaction mass was cooled to RT and ice cold water (20.0 ml) was added into it. Solid compound was formed and filtered to get compound 6 as an off white solid.
Spectral data of the synthesized compounds
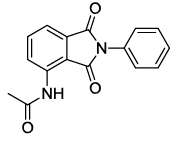
N-(1,3-dioxo-2-phenylisoindolin-4-yl)acetamide (6a)
Off white Solid, yield, 91%; M. p. 187-189oC, IR (KBr): 1736 cm-1 (strong, sharp, -HN-CO-), 1671 cm-1 (strong, broad, -CO- of amide carbonyl group); 1H-NMR (DMSO d6, 500 Mhz): δ (ppm)=2.20 (s, 3H, -CH3), 7.44 – 8.52 (m, 8H, Ar-H), 9.77 (s, 1H, -NH); 13C-NMR (DMSO d6, 125 MHz): δ (ppm)=169.2, 167.6, 166.4, 136.6, 135.8, 131.6, 131.5, 128.8, 128.1, 127.2, 125.7, 118.1, 116.9, 24.2; MASS: m/z=280.9 (M++1).
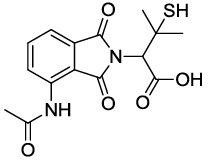
2-(4-acetamido-1,3-dioxoisoindolin-2-yl)-3-mercapto-3-methylbutanoic acid (6b)
Pale yellow Solid, yield, 89%; M. p. 186°C-188°C, IR (KBr): 1731 cm-1 (strong, sharp, -HN-CO-), 1668 cm-1 (strong, broad, -CO- of amide carbonyl group); 1H-NMR (DMSO d6, 500 MHz): δ (ppm)=1.43 (s, 3H, -CH3), 1.61 (s, 3H, -CH3), 2.20 (s, 3H, -CH3), 4.74 (s, 1H, -CH), 7.61-8.48 (m, 3H, Ar-H), 9.73 (s, 1H, -NH), 13.30 (s, 1H, -COOH); 13C-NMR (DMSO d6, 125 MHz): δ (ppm)=169.3, 167.9, 167.8, 166.9, 136.6, 136.1, 131.2, 126.3, 118.4, 116.7, 60.5, 45.8, 29.9, 29.7, 24.2; LC-MS: m/z=335.66 (M+-1).
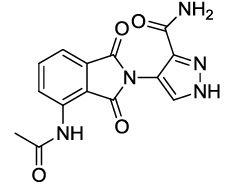
4-(4-acetamido-1,3-dioxoisoindolin-2-yl)-1H-pyrazole-3-carboxamide (6c)
Off white solid, yield, 85%; M. p. 192°C-194°C, IR (KBr): 3384 (Unequal doublet, asymmetric stretching of NH2), 1738 cm-1 (strong, sharp, -HN-CO-), 1660 cm-1 (strong, broad, -CO- of amide carbonyl group); 1H-NMR (DMSO d6, 500 MHz): δ (ppm)= 2.19 (s, 3H, -CH3), 7.37 (s, 2H, -NH2), 7.63 – 8.51 (m, 3H, Ar-H), 9.76 (s, 1H, -NH), 12.98 (s, 1H, -NH); 13C-NMR (DMSO d6, 125 MHz): δ (ppm)=169.8, 167.9, 166.9, 159.9, 137.2, 136.5, 136.1, 132.6, 130.6, 126.5, 121.8, 118.8, 118.1, 24.7; LC-MS: m/z=313.9 (M+-1).
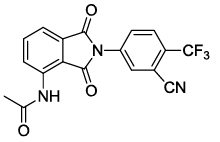
N-(2-(3-cyano-4-(trifluoromethyl)phenyl)-1,3-dioxoisoindolin-4-yl)acetamide (6d)
Off white solid; M. p. 202°C-204°C; IR (KBr): 2185 (sharp, medium, -CN), 1729 cm-1 (strong, sharp, -HN-CO-), 1664 cm-1 (strong, broad, -CO- of amide carbonyl group); 1H-NMR (DMSO d6, 500 MHz): δ (ppm)=2.22 (s, 3H, -CH3), 7.77- 8.52 (m, 6H, Ar-H), 9.79 (s, 1H, -NH); 13C-NMR (DMSO d6, 125 MHz): δ (ppm)=169.3, 166.5, 165.6, 136.9, 136.3, 131.6, 131.4, 131.2, 130.8, 126.4, 124.7, 123.3, 121.1, 118.6, 117.1, 115.2, 107.1, 24.2; LC-MS: m/z=372.60 (M+-1).
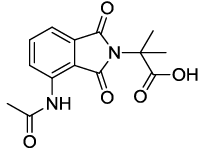
2-(4-acetamido-1,3-dioxoisoindolin-2-yl)-2-methylpropanoic acid (6e)
Off white solid; M. p. 200°C-202°C; IR (KBr): 1722 cm-1 (strong, sharp, -HN-CO-), 1658 cm-1 (strong, broad, -CO- of amide carbonyl group); 1H-NMR (DMSO d6, 500 MHz): δ (ppm)=1.72 (s, 6H, -CH3), 2.19 (s, 3H, -CH3), 7.51-8.47 (m, 3H, Ar-H), 9.71 (s, 1H, -NH) 12.91 (s, 1H, -COOH); 13C-NMR (DMSO d6, 125 MHz): δ (ppm)=173.7, 169.2, 168.8, 167.4, 136.3, 135.8, 131.3, 125.7, 117.7, 116.4, 59.8, 24.2; LC-MS: m/z=289.60 (M+-1).
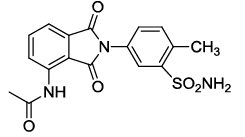
N-(2-(4-methyl-3-sulfamoylphenyl)-1,3-dioxoisoindolin-4-yl)acetamide (6f)
Pale brown solid; M. p. 212°C-214°C; IR (KBr): 3376 (Unequal doublet, asymmetric stretching of NH2),1719 cm-1 (strong, sharp, -HN-CO-), 1651 cm-1 (strong, broad, -CO- of amide carbonyl group); 1H-NMR (DMSO d6, 500 MHz): δ (ppm)=2.16 (s, 3H, -CH3), 2.47 (s, 3H, -CH3), 7.29 (s, 2H, -NH2), 7.55-8.41 (m, 6H, Ar-H), 9.68 (s, 1H, -NH); 13C-NMR (DMSO d6, 125 MHz): δ (ppm)=169.2, 167.4, 166.3, 142.5, 136.6, 135.9, 132.7, 131.7, 130.3, 129.3, 125.8, 118.2, 117.1, 24.2, 19.5; LC-MS: m/z=372.63 (M+-1).
N-(2-(4-cyclopropylnaphthalen-1-yl)-1,3-dioxoisoindolin-4-yl)acetamide (6g)
Brown solid; M. p. 208°C-210°C; IR (KBr): 1724 cm-1 (strong, sharp, -HN-CO-), 1662 cm-1 (strong, broad, -CO- of amide carbonyl group); 1H-NMR (DMSO d6, 500 MHz): δ (ppm)=2.11 (t, 2H, -CH2), 2.13 (t, 2H, -CH2), 2.18 (s, 3H, -CH3), 7.38 – 8.55 (m, 9H, Ar-H), 9.78 (s, 1H, -NH); 13C-NMR (DMSO d6, 125 MHz): δ (ppm)=169.2, 168.2, 167.2, 140.9, 136.7, 135.8, 133.4, 131.9, 130.1, 127.1, 126.8, 126.5, 125.9, 124.6, 124.4, 123.3, 122.7, 118.3, 117.4, 24.2, 12.9, 7.0, 6.8; LC-MS: m/z=371.0 (M++1).
N-(1,3-dioxo-2-(4H-1,2,4-triazol-4-yl)isoindolin-4-yl)acetamide (6h)
Pale brown Solid; M. p. 215°C-217°C; IR (KBr): 1720 cm-1 (strong, sharp, -HN-CO-), 1668 cm-1 (strong, broad, -CO- of amide carbonyl group); 1H-NMR (DMSO d6, 500 MHz): δ (ppm)=2.18 (s, 3H, -CH3), 7.77-8.43 (m, 3H, Ar-H), 8.88 (s, 2H, -CH), 9.90 (s, 1H, -NH); 13C-NMR (DMSO d6, 125 MHz): δ (ppm)=169.2, 162.8, 162.5, 143.1, 136.8, 136.6, 129.8, 127.9, 119.3, 116.1, 23.9; LC-MS: m/z=371.9 (M++1).
N-(2-(2-ethylphenyl)-1,3-dioxoisoindolin-4-yl)acetamide (6i)
Off white Solid; M. p. 217°C-218°C; IR (KBr): 1720 cm-1 (strong, sharp, -HN-CO-), 1667 cm-1 (strong, broad, -CO- of amide carbonyl group); 1H-NMR (DMSO d6, 500 MHz): δ (ppm)=1.08 (t, 3H, -CH3), 2.18 (s, 3H, -CH3), 2.48 (q, 2H, -CH2), 7.34-8.51 (m, 7H, Ar-H), 9.76 (s, 1H, - NH); 13C-NMR (DMSO d6, 125 MHz): δ (ppm)=169.2, 167.9, 166.9, 142.3, 136.7, 135.9, 131.8, 129.9, 129.6, 129.5, 129.0, 126.7, 126.1, 118.4, 117.3, 24.2, 23.8, 14.5; LC-MS: m/z=308.9 (M++1).
N-(2-(2-bromo-4-fluorophenyl)-1,3-dioxoisoindolin-4-yl)acetamide (6j)
Off white solid; M. p. 214°C-216°C; IR (KBr): 1726 cm-1 (strong, sharp, -HN-CO-), 1659 cm-1 (strong, broad, -CO- of amide carbonyl group); 1H-NMR (DMSO d6, 500 MHz): δ (ppm)=2.19 (s, 3H, -CH3), 7.46-8.52 (m, 6H, Ar-H), 9.80 (s, 1H, -NH); 13C-NMR (DMSO d6, 125 MHz): δ (ppm)=169.8, 167.2, 166.3, 161.6, 137.3, 136.7, 133.5, 132.1, 128.2, 127.1, 124.4, 120.7, 119.1, 117.7, 116.5, 24.7; LC-MS: m/z=376.9 (M++1).
N-(2-(1,3-dimethyl-1H-indazol-6-yl)-1,3-dioxoisoindolin-4-yl)acetamide (6k)
Pale brown solid; M. p. 219°C-221°C; IR (KBr): 1723 cm-1 (strong, sharp, -HN-CO-), 1648 cm-1 (strong, broad, -CO- of amide carbonyl group); 1H-NMR (DMSO d6, 500 MHz): δ (ppm)=2.20 (s, 3H, -CH3), 2.64 (s, 3H, -CH3), 4.08 (s, 3H, -NCH3), 6.98- 8.53 (m, 6H, Ar-H), 9.79 (s, 1H, - NH); 13C-NMR (DMSO d6, 125 MHz): δ (ppm)=169.2, 167.8, 166.7,146.2, 136.6, 135.8, 132.2, 131.8, 128.8, 125.7, 120.5, 119.9, 119.4, 118.1, 117.0, 115.6, 37.4, 24.2, 9.4; LC-MS: m/z=349.61 (M++1).
N-(2-(3-nitrophenyl)-1,3-dioxoisoindolin-4-yl)acetamide (6l)
Pale brown solid; M. p. 220°C-222°C; IR (KBr): 1731 cm-1 (strong, sharp, -HN-CO-), 1652 cm-1 (strong, broad, -CO- of amide carbonyl group); 1H-NMR (DMSO d6, 500 MHz): δ (ppm)=2.22 (s, 3H, -CH3), 7.68-8.53 (m, 7H, Ar-H), 9.79 (s, 1H, -NH); 13C-NMR (DMSO d6, 125 MHz): δ (ppm)=169.3, 167.0, 165.9, 147.8, 136.7, 136.1, 133.4, 132.6, 131.6, 130.3, 126.0, 122.7, 121.7, 118.4, 117.0, 24.2; LC-MS: m/z=325.2 (M++1).
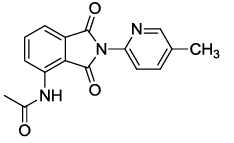
N-(2-(5-methylpyridin-2-yl)-1,3-dioxoisoindolin-4-yl)acetamide (6m)
Off white solid; M. p.224°C-226°C; IR (KBr): 1734 cm-1 (strong, sharp, -HN-CO-), 1662 cm-1 (strong, broad, -CO- of amide carbonyl group); 1H-NMR (DMSO d6, 500 MHz): δ (ppm)=2.19 (s, 3H, -CH3), 2.41 (s, 3H, -CH3), 7.37-8.51 (m, 6H, Ar-H), 9.78 (s, 1H, -NH); 13C-NMR (DMSO d6, 125 MHz): δ (ppm)=169.2, 167.0, 165.9, 149.7, 148.9, 145.6, 136.8, 136.1, 131.5, 126.1, 124.9, 123.4, 118.4, 116.9, 24.2, 20.4; LC-MS: m/z=396.1 (M++1).
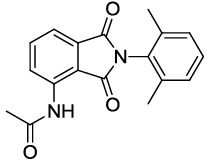
N-(2-(2,6-dimethylphenyl)-1,3-dioxoisoindolin-4-yl)acetamide (6n)
Off white Solid; M. p. 221°C-223°C; IR (KBr): 1731 cm-1 (strong, sharp, -HN-CO-), 1668 cm-1 (strong, broad, -CO- of amide carbonyl group); 1H-NMR (DMSO d6, 500 MHz): δ (ppm)=2.08 (s, 6H, -CH3), 2.19 (s, 3H, -CH3), 7.22-8.51 (m, 6H, Ar-H), 9.80 (s, 1H, -NH); 13C-NMR (DMSO d6, 125 MHz): δ (ppm)=169.2, 167.2, 166.3, 136.8, 136.7, 136.2, 131.4, 129.6, 129.3, 128.2, 126.4, 118.6, 117.1, 24.2, 17.5; LC-MS: m/z=309.1 (M++1).
Benzyl 2-(4-acetamido-1,3-dioxoisoindolin-2-yl)acetate (6o)
Pale brown solid; M. p. 221°C-223°C; IR (KBr): 1731 cm-1 (strong, sharp, -HN-CO-), 1654 cm-1 (strong, broad, -CO- of group), 1667 cm-1 (strong, broad, -CO- of amide carbonyl group); 1H-NMR (DMSO d6, 500 MHz): δ (ppm)=2.18 (s, 3H, -CH3), 4.49 (s, 2H, -CH2), 4.19 (s, 2H, - CH2), 7.34-8.43 (m, 8H, Ar-H), 9.75 (s, 1H, -NH); 13C-NMR (DMSO d6, 125 MHz): δ (ppm)=169.2, 167.5, 167.4, 166.6, 136.5, 136.1, 135.4, 131.6, 128.5, 128.3, 128.0, 126.5, 118.4, 117.2, 66.7, 38.7, 24.1; LC-MS: m/z=353.3 (M++1).
Pharmacology
Reagents and cell lines RAW 264.7 cells (murine macrophage cell line) were procured from ATCC (Washington D.C., USA) and usually maintained in RPMI 1640 medium with 10% fetal bovine serum.Mouse TNF-α ELISA kit was purchased from R&D Systems (Minneapolis, MN, USA). Lipopolysaccharide (LPS) was obtained from Escherichia coli strain 0127: B8 purchased from Sigma-aldrich (St. Louis, MO, USA).
TNF-α production assay
TNF-α production was measured after few modifications following a procedure described in the literature [27]. RAW 264.7 cells were pre-incubated either with compound or DMSO (vehicle control) for 30 minutes and then stimulated with 1 μg/ml of LPS overnight. Preliminary screening of the compounds was performed at 30 μM and dose response studies were carried out at eight different concentrations (30, 10, 3, 1, 0.3, 0.1, 0.03, 0.01 μM). Poststimulation, cell supernatants were harvested, centrifuged to clear cell debris and the amount of TNF-α in the supernatants was measured using mouse TNF-α DuoSet ELISA kit from R&D Systems according to manufacturer’s recommendations. The percentage of inhibition was calculated using the following formula:
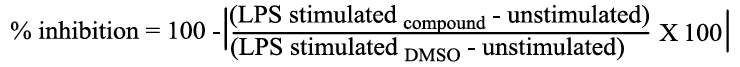
A nonlinear regression analysis was performed to determine the IC50 values from dose response curve. IC50 values are expressed as mean ± SD.
Results and Discussions
To optimize and establish the reaction conditions we started the synthesis by taking N-(1,3-dioxo-2-phenylisoindolin-4-yl)acetamide as a model reaction. We initiated the synthesis of N-(1,3-dioxo-1,3- dihydroisobenzofuran-4-yl-acetamide as a key starting material to synthesize N-(1,3- dioxo-2-phenylisoindolin-4-yl)acetamide. As shown in Scheme 1, 3-nitropthalic acid 1 was reduced with hydrazine hydrate 2 to form 3- aminopthalic acid 3 in presence of FeCl3.6H2O at 70°C-75°C for 1-2 h. Compound 3 on further reaction with acetic anhydride under reflux conditions for 2-3 h yielded the key starting material N-(1,3-dioxo-1,3-dihydroisobenzofuran-4-yl-acetamide 4. The structure of the product was confirmed by using IR, NMR & Mass spectra.
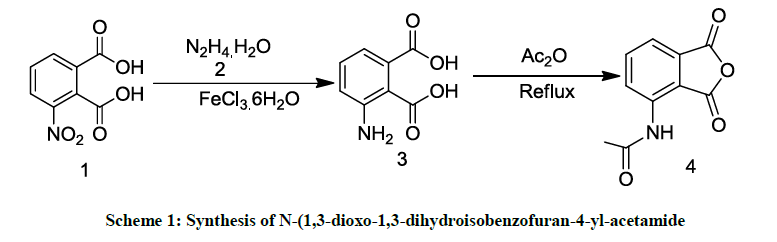
Scheme 1: Synthesis of N-(1,3-dioxo-1,3-dihydroisobenzofuran-4-yl-acetamide
After synthesizing compound 4 we want to examine the main condensation reaction between compound 4 and aniline 5a using different solvents to get N-(1,3-dioxo-2-phenylisoindolin-4-yl)acetamide (Scheme 2). To examine the role of solvent we performed the reaction in different solvents. The results were summarized in Table 1. When a mixture of compound 4 and aniline 5a was stirred at reflux using methanol as solvent for 60 min the desired compound 6a was formed in 71% yield (Table 1, entry 1). The use of other organic solvents did not improve the product 6a yield. All the reactions proceeded well but only moderate yields were obtained (Table 1, entries 2-7). Then we shifted our focus to improve the yield by using eco-friendly solvents such as water, acetic acid, polyethylene glycol, ethanol, ethylene glycol, glycerol for this condensation reaction.
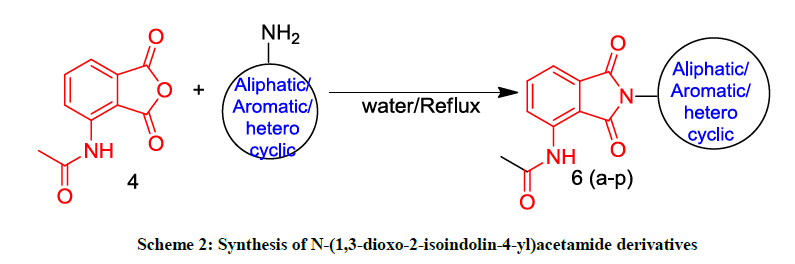
Scheme 2: Synthesis of N-(1,3-dioxo-2-isoindolin-4-yl)acetamide derivatives
| S. No. | Solvent | Time (min)/ Temp | Temperature | Yield (%) |
|---|---|---|---|---|
| 1 | Methanol | 60 | Reflux | 71 |
| 2 | DMF | 80 | 1100°C | 68 |
| 3 | THF | 80 | Reflux | 68 |
| 4 | CHCl3 | 100 | Reflux | 59 |
| 5 | 1,4-dioxane | 80 | Reflux | 67 |
| 6 | acetone | 80 | Reflux | 62 |
| 7 | acetonitrile | 80 | Reflux | 65 |
Table 1: Effect of organic solvents for the formation of 6a
When a mixture of 4a (1 mmol) and aniline 5a was stirred in water at room temperature for 10 h only a trace amount of desired product (6a) was obtained (Table 2, entry 1 ). Surprisingly, the yield of 6a was increased drastically to 94% when the same reaction was performed in water for 60 min at reflux (Table 2, entry 2).
| S. No. | Solvent | Temperature (°C) | Time (Min) | Yield (%) |
|---|---|---|---|---|
| 1 | Water | RT | 600 | 5 |
| 2 | Water | Reflux | 60 | 94 |
| 3 | Water | Reflux | 90 | 94 |
| 4 | Acetic acid | Reflux | 60 | 89 |
| 5 | Ethanol | Reflux | 60 | 87 |
| 6 | Ethylene glycol | Reflux | 60 | 90 |
| 7 | PEG-600 | Reflux | 60 | 88 |
| 8 | PEG-400 | Reflux | 60 | 88 |
| 9 | Glycerol | Reflux | 60 | 86 |
| 10 | PPA | Reflux | 60 | 84 |
aIsolated yield
Table 2: Effect of green solvents for the formation of 6aa
The use of other ecofriendly solvents, for example, acetic acid, polyethylene glycol, ethanol, ethylene glycol, glycerol were examined and found to be effective (Table 2, entries 4–10). Finally, the best results were obtained when the reaction was performed using water as solvent (Table 2, entry 2).
The increase in the duration of the reaction kept in water did not improve the product yield whereas decrease in temperature lowered the yield (Table 1, entries 1-2). After screening both the organic and ecofriendly solvents it was evident that the reaction proceeds smoothly in water for 60 minutes under reflux conditions.
To understand the malleability of the current methodology, a wide range of aromatic, aliphatic and heterocyclic amines were used with compound 4a in water. The results were presented in Table 3. In this methodology aromatic amines with electron-withdrawing or electron-donating substituents along with aliphatic and heterocyclic amines gave good yields (81%-94%). From the results it was evident that a variety of substituents on aromatic and heterocyclic rings were well tolerated. All the synthesized compounds were characterized by using IR, NMR and Mass spectra.
| S. No. | Aniline 5(a-o) | Product 6(a-o) | Yield (%) |
|---|---|---|---|
| 1 |  5a 5a |
 6a |
94 |
| 2 | 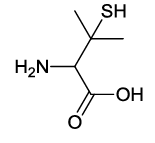 5b 5b |
 6b |
89 |
| 3 | 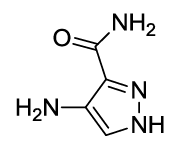 5c 5c |
 6c |
85 |
| 4 | 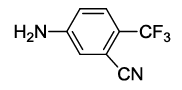 5d 5d |
 6d |
88 |
| 5 | 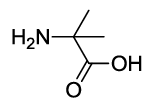 5e 5e |
 6e |
86 |
| 6 | 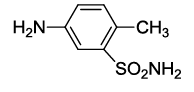 5f
5f |
 6f |
82 |
| 7 | 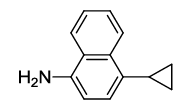 5g 5g |
 6g |
84 |
| 8 |  5h
5h |
 6h |
87 |
| 9 |  5i
5i |
 6i |
81 |
| 10 |  5j 5j |
 6j |
83 |
| 11 |  5k |
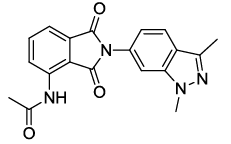 6k |
87 |
| 12 | 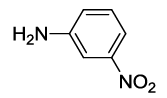 5l |
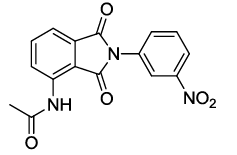 6l |
83 |
| 13 | 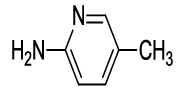 5m 5m |
 6m |
85 |
| 14 |  5n 5n |
 6n |
85 |
| 15 |  5o 5o |
 6o |
83 |
aisolated yields
Table 3: The synthesis of substituted 1, 3-dioxoisoindolin derivatives 6(a-q)a
All the synthesized compounds were tested for their TNF- α inhibitory potential in vitro [28]. Compounds 6d, 6e, 6h, 6k, and 6o showed significant inhibition of TNF- α at 30 μM (Figure 1) and dose response studies were carried out at eight different concentrations (30, 10, 3, 1, 0.3, 0.1, 0.03, 0.01 μM). Whereas the compound 6d showed dose-depended inhibition with an IC50 value 8.76 μM.
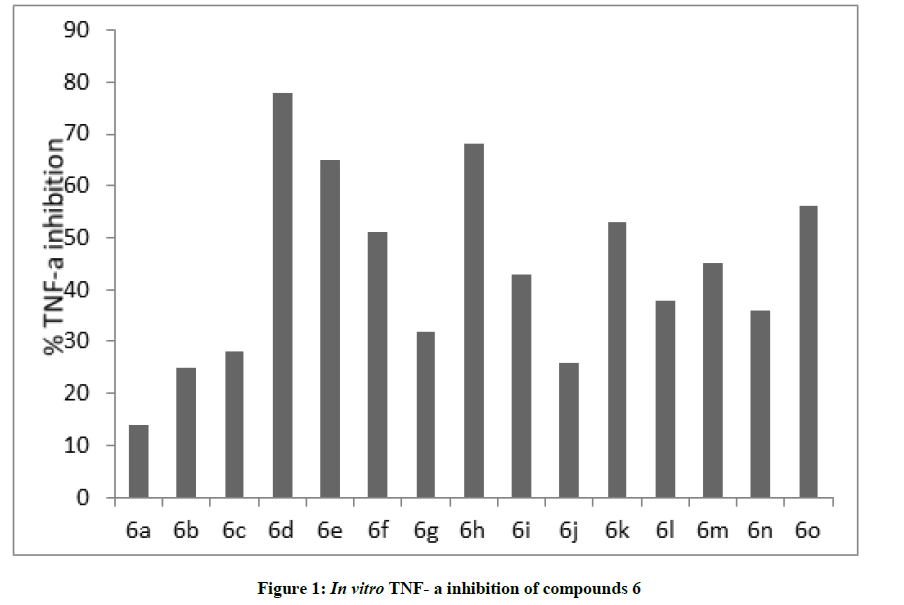
Figure 1: In vitro TNF- a inhibition of compounds 6
This was supported by the docking results of 6d with TNF-α protein which showed strong interactions with the hydrophobic binding site (binding energy 8.78 Kcal/mol) consisting primarily of serine, leucine and tyrosine residues. Docking interactions showed that 6d showed docking with binding pocket formed of Ser60, Trp61, Phe64, Lys65, Ile83, Asp130,Tyr56, Leu57,Tyr59, Val62, Arg82, Tyr119, Leu120, Gly121, Leu132, Ser133, Ala134, Ile136 (Figure 2). Thus, compounds 6a–o may have medicinal value with potential therapeutic applications.
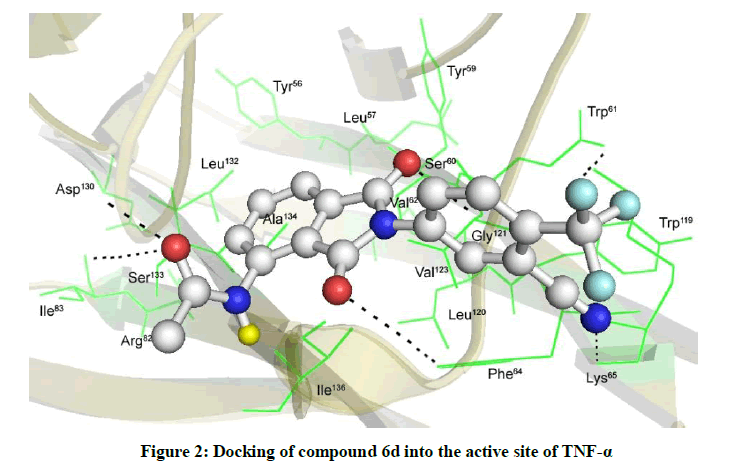
Figure 2: Docking of compound 6d into the active site of TNF-α
Conclusion
In summary, green and eco-friendly synthetic method have been developed for the synthesis of N-(1,3-dioxo-2-phenylisoindolin-4-yl)acetamide derivatives 6(a-q) under catalyst free conditions. This new method has the advantages of higher yields, mild reaction conditions, shorter reaction times, convenient procedure and environmental friendliness. This research has directed to the identification of new potent inhibitor of TNF-α.
Acknowledgement
Authors are very thankful to the authorities of Granules India pvt. ltd and Jawaharlal Nehru Technological University Hyderabad for providing laboratory facilities and financial support.
References
- J.B. Bartlett, K. Dredge, A.G. Dalgleish, Nat. Rev., 2004, 4, 314.
- J.D. Klausner, V.H. Freedman, G. Kaplan, Clin. Immunol. Immunopathol., 1996, 81, 219.
- A.L. Machado, L.M. Lima, J.X. Araújo jr, C.A.M. Fraga, V.L.G. Koatz, E.J. Barreiro, Bioorg. Med. Chem. Lett., 2005, 15, 1169.
- L. Mazzoccoli, S.H. Cadoso, G.W. Amarante, M.V.N. Souza, R. Domingues, M.A. Machado, M.V. Almeida, H.C. Teixeira, Biomed. Pharmacother., 2012, 66, 323-329.
- P. Andrade, V. Visser-vandewalle, J.S. Del rosario, M.A. Daemen, W.A. Buurman, H.W. Steinbusch, G. Hoogland, Brain Res.,2012, 1450, 24-32.
- G. Palencia, C. Rubio, V. Custodio-ramirez, C. Paz, J. Sotelo, Epilepsy Res., 2011, 95, 263.
- C. Pessoa, P.M. Ferreira, L.V. Lotufo, M.O. de Moraes MO, S.M. Cavalcanti SM, L.C. Coêlho LC, M.Z. Hernandes MZ, A.C. Leite AC, C.A. De Simone CA, V.M. Costa VM, V.M. Souza VM, 2010, 5, 523.
- B. Badamtseren, E. Odkhuu, N. Koide, A. Haque, Y. Naiki, S. Hashimoto, T. Komatsu, T. Yoshida, T. Yokochi, Cell Immunol., 2011, 270, 19.
- C. Nabhan, D.P. Petrylak, Cancer Clin. Genitourinary Cancer., 2012, 10, 141.
- Y. Kagoya, Y. Nannya, M. Kurokawa, Leukemia Res., 2012, 36, 1016.
- M.O. Simon, C.J. Li, Chem. Soc. Rev., 2012, 41, 1415.
- A.S. Roger, Green Chem., 2005, 7, 267.
- B.M. Gawande, D.B.V. Bonifácio, S.R.L. Paula Branco, S. Rajender Varma, Chem. Soc. Rev., 2013, 42, 5522.
- H.R. Safaei, M. Shekouhy, S. Rahmanpur, A. Shirinfeshan, Green Chem.,2012, 14, 1696.
- J. Chen, S.K. Spear, J.G. Huddleston, R.D. Rogers, Green Chem., 2005, 7, 64.
- N.V. Plechkova, K.R. Seddon, Chem. Soc. Rev.,2008, 37, 123.
- S. Shafi, M.M. Alam, N. Mulakayala, C. Mulakayala, G. Vanaja, A.M. Kalle, R. Pallu, M.S. Alam, Eur. J. Med. Chem., 2012, 49, 324.
- N. Mulakayala, P. Rao, J. Iqbal, R. Bandichhor, S. Oruganti, Eur. J. Med. Chem., 2013, 60, 170.
- H. Sudhakar, G. Pavana Kumari, N. Mulakayala, Ind. J. Adv. Chem. Sci., 2013, 2, 57.
- H. Sudhakar, G. Pavana Kumari, R. Venkata Nadh, N. Mulakayala, Ind. J. Adv. Chem. Sci., 2014, 2, 294.
- Ismail, B. Kuthati, G. Thalari, V. Bommarapu, C. Mulakayala, S.K. Chita, N. Mulakayala, Bioorg. Med. Chem. Lett., 2017, 27, 1446.
- N. Mulakayala, B. Kandagatla, Ismail, R.K. Rapolu, P. Rao, C. Mulakayala, C.S. Kumar, J. Iqbal, S. Oruganti, Bioorg. Med. Chem. Lett., 2012, 15, 5335.
- N. Mulakayala, D. Rambabu, M.R. Raja, M. Chaitanya, C.S. Kumar, A.M. Kalle, G.R. Krishna, C.M. Reddy, M.V.B. Rao, M. Pal, Bioorg. Med. Chem., 2012, 20, 759.
- R.M. Rao, Ch.U. Reddy, A. Nakhi, N. Mulakayala, M. Alvala, M.K. Arunasree, R. Poondra, J. Iqbal, M. Pal, Org. Biomol. Chem., 2011, 9, 3808.
- K. Manjulatha, S. Srinivas, N. Mulakayala, D. Rambabu, M. Prabhakar, K.M.Arunasree, M. Pal, Bioorg. Med. Chem lett., 2012, 22, 6160.
- M.R. Gudisela, N. Srinivasu, C. Mulakayala, P. Bommu, M.V. Basaveswara Rao, M. Naveen, Bioorg. Med. Chem lett., 2017, 27, 4140.
- Bruker, SADABS V2008-1, Bruker AXS.: Madison, WI, USA, 2008.
- K.V. Parsa, L.P. Ganesan, M.V. Rajaram, M.A. Gavrilin, A. Balagopal, N.P. Mohapatra, M.D. Wewers, L.S. Schlesinger, J.S. Gunn, S. Tridandapani, PLoS Pathog., 2006, 2, e71.



