Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 6
Biological Screening for a New Series of Thiophene/Pentahydrocycloheptathieno[2,3-D]Pyrimidine Derivatives along with their Synthetic Strategy
1Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Helwan University, Ain Helwan, PO Box 11795, Cairo, Egypt
2Department of Pharmaceutical Sciences, Faculty of Pharmacy, Princess Nourah bint Abdelrahman University, PO Box 84428, Riyadh, Saudi Arabia
- *Corresponding Author:
- Elshaymaa I Elmongy
Department of Pharmaceutical Chemistry
Faculty of Pharmacy
Helwan University
Ain Helwan, PO Box 11795, Cairo, Egypt
Abstract
This work describes anticancer and antioxidant biological testing for new derived compounds form cycloheptathienopyrimidine nucleus along with their synthetic approach. The anticancer activity was tested against cancerous human breast cells MCF-7 and human liver cancer cell line HEPG-2 in reference to Doxorubicin. All the tested products showed significant cytotoxic activity. Six out of twelve tested compounds showed anticancer activity even more than that of reference standard Doxorubicin against MCF-7 cell line whereas, eight compounds declared cytotoxic effect more than the reference standard against cell line HEPG-2 reflected by their IC50S. Furthermore, six of the top ranked anticancer results were tested for their antioxidant effect with DPPH method in reference to Ascorbic acid, three of which gave 100% free radical scavenging activity.
Keywords
Thienopyrimidine, Thiophene, Sulfonamides, Anticancer, Antioxidant
Introduction
Thienopyrimidines are considered structural analogues of biogenic purine, they are endowed with variety of biological activities (Figure 1) and have been employed extensively as a scaffold in the design of compounds in agrochemical industry [1-3] and versatile biologically active compounds as enzyme inhibitors including Aurora kinase, 6 tyrosine kinases [3,4], cyclin-dependent kinase [5], Polyadp Ribose Polymerase (PARP) inhibitors [6], antifungal [7], antiviralagents [1], anticancer agents against some tumor cell lines [8-12] as colorectal cancerous cell (HCT116), Hepatic adenoma (HEPG-2), Chronic myelogenous leukemia (CML), cancerous breast cell (MCF-7) in addition to their reported antioxidant activity [13] which is known to be effective not only in prophylaxis but also in treatment of complicated diseases as Alzheimer, cancer, and stroke. Many of these inhibitors are currently under pre-clinical or clinical trials for their treatment of neurodegenerative, autoimmune, inflammatory diseases and cancer [14].
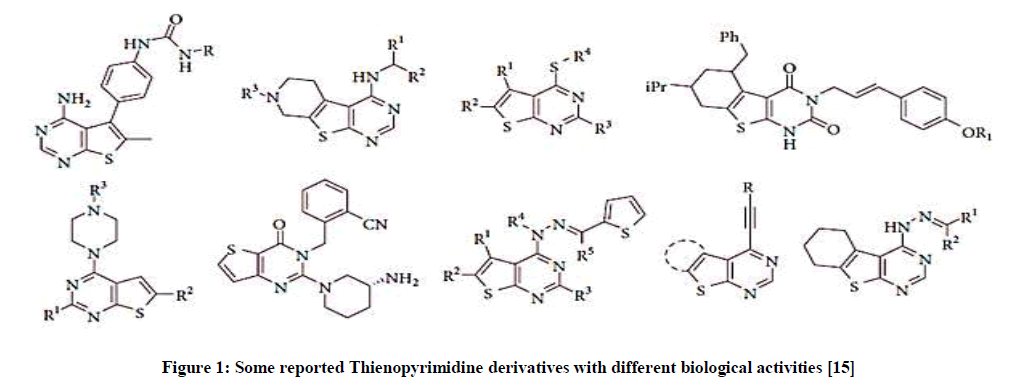
Figure 1: Some reported Thienopyrimidine derivatives with different biological activities [15]
The aforementioned facts provoked our interest to synthesize a series of thiophenes and thienopyrimidines (Figure 2) aiming to obtain new compounds with antioxidant and anticancer activities, taking into consideration that the anticancer activity in compounds bearing the sulfonamide moiety may be due to carbonic anhydrase enzyme inhibition (Figure 3).
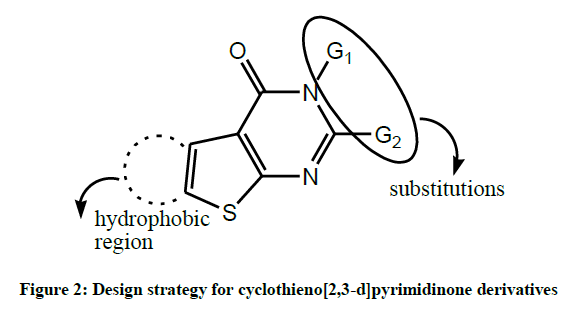
Figure 2: Design strategy for cyclothieno[2,3-d]pyrimidinone derivatives
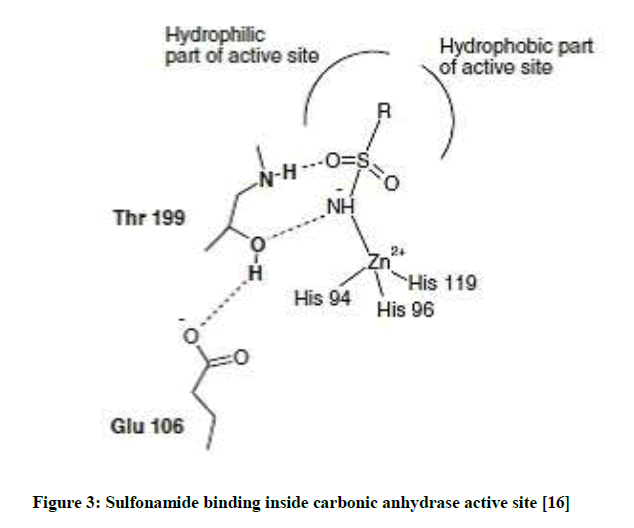
Figure 3: Sulfonamide binding inside carbonic anhydrase active site [16]
Materials and Methods
Chemistry
Materials
Infrared analysis was performed on Bruker FT-IR spectrophotometer using KBr discs, at the Micro-analytical center, faculty of Science, Cairo University, Cairo, Egypt and were expressed as wave number (cm-1). The 1H-NMR analysis was performed at the Faculty of Science Micro-analytical center, Cairo University, Cairo, Egypt on Varian Mercury VX-300 NMR spectrophotometer at 300 MHZ. The (13C-NMR) Nuclear Magnetic Resonance was done on apparatus Varian Mercury VX-300 NMR spectrophotometer at 75.446 MHZ using (DMSO-d6), at main defense chemical laboratories, Cairo, Egypt. Elemental analyses were detected at Al-Azhar University Laboratory of the Regional Center for Mycology and Biotechnology. Melting points (M.p.) were recorded on electro thermal IA9100 apparatus (Shimadzu, Japan). Compounds 1 and 2 were prepared according to the reported procedures [17-19] respectively.
General method (3 a-c)
A mixture of 2 (2.81 g, 0.01 mol) and the suitable sulfonamide (0.01 mol) in dioxane (20 ml), was refluxed for 6 h. The formed solid was filtered while hot using ethanol for crystallization to give 3 a-c.
Ethyl-2-(3-(p-sulfamoylphenyl)thioureido)-4,5,6,7,8-pentahydrocyclohepta[b]thiophene-3-carboxylate (3a): Yield 92%, M.p. 200-202°C; IR (KBr disc) (cm-1) 3472, 3380, 3350 (2NH, NH2), 1702 (C=O); Anal. for C19H23N3O4S3 (453.599); Calcd %, C, 50.31; H, 5.11; N, 9.26. Found C, 50.35; H, 5.06; N, 9.11; 1H-NMR (DMSO) δ (ppm)=1.22 (t, 3H, CH3), 1.34-2.97 (m, 10H cycloheptane) 4.22 (q, 2H, CH2―CH3), 7.12-7.49 (m, 4H, aromatic), 6.74 (s, 2H, NH2, D2O interchangeable), 10.83 (s, 1H, NH, D2O replaceable), 11.50 (s, 1H, NH, exchanged with D2O); 13C-NMR (DMSO-d6) δ (ppm)=14.29, 26.53, 27.41, 27.79, 27.90, 31.50, 60.45, 113.87, 123.53, 124.23, 128.87, 128.33, 136.81, 138.21, 140.23, 145.54, 160.21, 164.13, 175.64.
Ethyl-2-(3-(4-(pyrimidin-2-yl-sulfamoyl)phenyl)thioureido)-4,5,6,7,8-pentahydrocyclohepta [b]thiophene-3-carboxylate (3b): Yield 86%, M.p. 209-211°C; IR (cm-1) 3406, 3292 (NH), 1710 (C=O), 1582 (C=N); Anal. for C23H25N5O4S3 (531.671) Calcd %, C, 51.96; H, 4.74; N, 13.17. Found C, 52.02; H, 4.77; N, 13.13; 1H-NMR (DMSO-d6): 1.26-2.97 (m, 13H, 3H-CH3 and 10H cycloheptane) 4.19 (q, 2H, CH2―CH3), 7.12–8.18 (m, 7H, (3H pyrimidine and 4H aromatic)), 10.92 (s, 1H, NH, D2O replaceable), 11.51 (s, 1H, NH, D2O interchangeable), 12.03 (s, 2H, NH, D2O replaceable); 13C-NMR (DMSO-d6) δ (ppm)=14.22, 26.41, 27.11, 27.80, 28.21, 32.13, 60.01, 110.31, 116.23, 123.53, 126.23, 128.00, 132.24, 138.21, 140.42, 145.54, 148.23, 157.37, 158.34, 168.33, 169.30, 174.82.
Ethyl-2-(3-(4-methylpyrimidin-2-yl-sulfamoyl)phenyl)thioureido)-4,5,6,7,8-pentahydrocyclohepta [b]thiophene-3-carboxylate (3c): Yield 86%, M.p. 209-211°C; IR (KBr) (cm-1) 3488, 3382 (NH), 1702 ( C=O), 1592 (C=N); Anal. for C24H27N5O4S3 (545.697) Calcd %, C, 52.82; H, 4.99; N, 12.83. Found C, 52.66; H, 5.21; N, 12.78; 1H-NMR (DMSO-d6): 1.26-2.97 (m, 16H, 3H (CH3), 3H (CH3) and 10H cycloheptane), 4.19 (q, 2H, CH2―CH3), 6.51(m, 2H, pyrimidine), 7.60-8.18 (m, 4H aromatic), 10.89 (s, 1H, NH, D2O replaceable), 11.32 (s, 1H, NH, D2O replaceable), 11.88 (s, 1H, NH, D2O interchangeable); 13C-NMR (DMSO-d6) δ (ppm)=14.34, 24.80, 28.35, 27.81, 28.30, 28.94, 32.55, 60.15, 108.88, 116.47, 124.43, 124.96, 128.07, 128.33, 138.81, 140.10, 142.63, 145.54, 156.64, 160.21, 164.13, 168.78, 170.96, 179.44.
General method (4a-c)
A blend of (0.01 mol) of the appropriate compounds 3a-c and hydrazine hydrate (0.05 ml, 0.01 mol) in ethanol (20 ml), was refluxed for 6 h. The resulted solid was gathered by filtration while hot using dioxane for crystallization to yield 4a-c.
Amino-2-(4-sulfamoylphenylamino)-5,6,7,8,9-pentahydrocyclohepta[4,5]thieno–[2,3-d] pyrimidin-4(3H)-one (4a): Yield 62%, M.p. 251- 253°C; IR (KBr) (cm-1): 3439-3311 (NH2, NH), 1680 (C=O), 1336, 1148 (SO2); Anal. for C17H19N5O3S2 (405.494) Calcd %, C, 50.35; H, 4.72; N, 17.27. Found C, 50.31; H, 4.69; N, 17.34; 1H-NMR (DMSO-d6): 1.29-2.36 (m, 10H of cycloheptane ), 4.60 (s, NH2 D2O interchangeable), 5.41 (s, NH, D2O interchangeable), 7.16-7.49 (m, 4H of aromatic ring), 13.85 (s, NH D2O replaceable); 13C-NMR (DMSO-d6) δ (ppm)=26.78, 27.19, 27.23, 28.94, 31.87, 117.12, 119.24, 128.42, 129.63, 136.55, 141.20, 144.31, 150.53, 155.49, 157.37, 161.13, 164.84.
3-Amino-2-(4-[(pyrimidin-2-yl)sulfamoyl]-phenyl}amino)-5,6,7,8,9 pentahydrocyclohepta[4,5]thieno[2,3-d]pyrimidin-4(3H)-one (4b): Yield 67%, M.p. 248-250°C; IR (KBr disc) (cm-1): 3410-3380 (NH2, NH), 1679 (C=O), 1332, 1152 (SO2); 1H-NMR (DMSO-d6): 1.29-2.36 (m, 10H of cycloheptane), 4.11 (s, NH2, exchanged with D2O), 4.78 (s, NH, D2O interchangeable), 6.98-7.40 (m, 3H of pyrimidine and 4H of aromatic ring), 11.65 (s, NH, exchanged with D2O); Anal. for C21H21N7O3S2 (483.567) Calcd %, C, 52.16; H, 4.38; N, 20.28. Found C, 52.01; H, 4.49; N, 20.13; 13C-NMR (DMSO-d6) δ (ppm)=25.76, 27.04, 27.63, 28.94, 32.12, 108.34 117.44, 119.24, 128.63, 129.51, 135.89, 141.20, 145.01, 150.53, 155.49, 157.37, 157.91, 158.20, 161.13, 164.84, 169.44.
3-Amino-2-(4-[(4-methylpyrimidin-2-yl)sulfamoyl]phenyl}amino)-5,6,7,8,9-pentahydrocyclohepta[4,5]thieno–[2,3-d] pyrimidin-4(3H)- one (4c): Yield 78%, M.p. 230-232°C; IR (KBr disc) (cm-1): 3430-3390 (NH2, NH), 1682 (C=O), 1338, 1152 (SO2); Anal. for C22H23N7O3S2 (497.593) Calcd %, C, 53.10; H, 4.66; N, 19.70. Found C, 53.28; H, 4.59; N, 19.83; 1H-NMR (DMSO-d6): 1.22-2.32 (m, 10H of cycloheptane ), 2.35 (s, 3H, CH3 pyrimidine), 4.41 (s, NH2, exchanged with D2O), 5.20 (s, NH, D2O replaceable), 7.00-7.77 (m, 2H of pyrimidine and 4H of aromatic ring), 12.15 (s, NH D2O interchangeable); 13C-NMR (DMSO-d6) δ (ppm)=23.82, 25.83, 27.04, 27.32, 28.46, 32.12, 108.34, 116.00, 128.42, 129.45, 137.86, 140.11, 148.31, 150.40, 156.49, 157.37, 158.91, 160.20, 161.13, 166.72, 168.01.
General method (5a, b)
A solution of the proper substituted-4-sulfamoylphenylamino-5,6,7,8,9-entahydrocyclohepta[4,5]thieno[2,3-d]pyrimidinone derivatives 4b and 4c (0.01 mol) reacted with formic acid (20 ml), upon reflux for 8h. The product was then concentrated, crystallized using ethanol to give 5a and 5b respectively.
3-{4-[2-pyrimidinylsulfamoyl]phenyl}-6,7,8,9,10-pentahydrocyclohepta[4,5]thieno[2,3-d]-1,2,4-triazolo[1,5-a]pyrimidin-11-one (5a): Yield 77%, M.p. 223-225°C; IR (KBr disc) (cm-1): 3400 (NH), 1695 (C=O), 1410, 1160 (SO2); Anal. for C23H21N7O3S2 (493.561) Calcd %, C, 53.54; H, 3.88; N, 19.87. Found C, 53.43; H, 3.91; N, 19.79; 1H-NMR (DMSO-d6): 1.37-2.55 (m,10H of cycloheptane), 6.70 (s, CH, triazol), 7.30-7.87 (m, 3H of pyrimidine and 4H of aromatic ring), 12.00 (s, NH, D2O replaceable); 13C-NMR (DMSO-d6) δ (ppm)=19.70, 24.63, 29.21, 29.36, 31.62, 110.30, 116.43, 117.02, 117.89, 125.62, 128.81, 129.33, 139.30, 146.80, 156.20, 157.94, 160.41, 163.36, 169.61.
3-{4-[(4-methylpyrimidin-2-yl)sulfamoyl]phenyl}-6,7,8,9,10-pentahydrocyclohepta[4,5]thieno[2,3-d]-1,2,4-triazolo[1,5-a]pyrimidin-11- one (5b): Yield 74%, M.p. 215-217°C; IR (KBr disc) (cm-1): 3410 (NH), 1678 (C=O), 1390, 1110 (SO2); Anal. for C23H21N7O3S2 (507.588) Calcd %, C, 54.42; H, 4.17; N, 19.32. Found C, 54.46; H, 4.13; N, 19.35; 1H-NMR (DMSO-d6): 1.91-2.83(m, 3H, CH3 and 10H of cycloheptane), 6.77 (s, CH, triazol), 7.11-7.64 (m, 2H of pyrimidine and 4H of aromatic ring), 12.34 (s, NH, D2O interchangeable).
General method (6a, b)
A solution of substituted-4-sulfamoylphenylamino-5,6,7,8,9-pentahydrocyclohepta[4,5]thieno–[2,3-d] pyrimidinone derivatives 4b and 4c (0.01 mol) and acetic anhydride (20 ml), was refluxed for 8 h. The formed product was dissipated then crystallized using ethanol to yield 6a and 6b respectively.
2-Methyl-3-{4-[2-pyrimidinylsulfamoyl]phenyl}-6,7,8,9,10-pentahydrocyclohepta[4,5]thieno[2,3-d]-1,2,4-triazolo[1,5-a]pyrimidin-11-one (6a): Yield 76%, M.p. 145-147°C; IR (KBr disc) (cm-1): 3231 (NH), 3100 (CH aromatic), 2986, 2934 (CH aliphatic), 1702 (C=O), 1588 (C=N), 1390, 1110 (SO2); Anal. for C23H21N7O3S2 (507.588) Calcd %, C, 54.42; H, 4.17; N, 19.32. Found C, 54.66; H, 4.25; N, 19.50; 1H-NMR (DMSO-d6): 1.22-2.23 (m, 10H of cycloheptane), 2.33 (s, CH3 of triazol), 7.20-7.61 (m, 3H of pyrimidine and 4H aromatic), 12.28 (s, NH, D2O interchangeable).
2-methyl-3-{4-[(4-methylpyrimidin-2-yl)sulfamoyl]phenyl}-6,7,8,9,10-pentahydrocyclohepta[4,5]thieno[2,3-d]-1,2,4-triazolo[1,5- a]pyrimidin-11-one (6b): Yield 74%, M.p. 123-125°C; IR (KBr disc) (cm-1): 3329 (NH), 3100 (CH aromatic), 2981, 2931 (CH aliphatic), 1672 (C=O), 1549 (C=N), 1375, 1157 (SO2); Anal. for C24H23N7O3S2 (521.615) Calcd %, C, 55.26; H, 4.44; N, 18.80. Found C, 55.19; H, 4.61; N, 18.77; 1H-NMR (DMSO-d6): 2.09-3.33 (m, 3H, CH3 of pyrimidine; 3H,CH3 of triazol and 10H of cycloheptane ), 7.14-7.97 (m, 2H of pyrimidine and 4H of aromatic ring), 13.11 (s, NH, D2O replaceable).
3-Amino-2-thioxo-5,6,7,8,9-pentahydrocyclohepta[4,5]thieno[2,3-d]pyrimidin-4(1H)one (7): Equivalent amounts(0.05 mol) of the isothiocyanate derivative 2 and hydrazine hydrate were refluxed for 6 h in ethanol. Precipitated upon cooling then gathered after filteration to be washed with ethanol and treated with 10% HCl to yield compound 7. Yield 67%, M.p. 176-177°C; IR (KBr disc) (cm-1): 3940 (NH), 2893 (CH aliphatic), 1659 (C=O); Anal. for C11H13N3OS2 (267.37) Calcd %, C, 49.41; H, 4.90; N, 15.72. Found C, 49.52; H, 4.88; N, 15.78; 1H-NMR (DMSO-d6): 1.29-3.40 (m, 10H of cycloheptane), 12.23 (s, NH), 13.21 (s, NH2); 13C-NMR (DMSO-d6) δ (ppm)=26.59-31.67 (5C-cycloheptane), 116.90, 125.22, 139.11, 157.37, 161.40, 172.46.
2-anilino-6,7,8,9,10-pentahydrocyclohepta[4,5]thieno[2,3-d]-1,3,4-thiadiazolo[3,2-a]pyrimidin-11-one (8): Equimolar amounts (0.01) of the 3-Amino-2-thioxo-thienopyrimidine derivative 7 and phenyl isothiocyanate were refluxed for 10 h in pyridine (20 ml). The formed mixture was then poured after cooling onto ice water mixture, the formed solid was collected by filtration before using methanol for crystallizationto give 8.
Yield 69%, M.p. 152-155°C; IR (KBr disc) (cm-1): 3390 (NH), 3080 (CH aromatic), 2960 (CH aliphatic), 1700 (C=O), 1590(C=N); Anal. for C18H16N4OS2 (368.492) Calcd %, C, 58.69; H, 4.38; N, 15.21. Found C, 58.72; H, 4.52; N, 15.19; 1H-NMR (DMSO-d6): 1.34-2.97 (m, 10H cycloheptane), 12.23 (s, NH), 7.16-7.49 (m, 5H of aromatic ring); 13C-NMR (DMSO-d6) δ (ppm)=20.59-31.67 (5C- cycloheptane), 116.31, 117.41, 118.88, 125.20, 129.60, 139.31, 144.12, 155.22, 158.10, 160.13, 163.66.
2-methyl-6,7,8,9,10-pentahydrocyclohepta[4,5]thieno[2,3-d]-1,3,4-thiadiazolo[3,2-a]pyrimidin-11-one (9): A blend of 7 (2.67 g, 0.01 mol) and acetic anhydride (10 ml) was stirred with reflux for 16 h. The reaction mixture was evaporated to give a sticky paste which was triturated by ethanol. The formed solid was filtered and crystallized upon treatment with dioxane to give 9. Yield 67%, M.p. 126-128°C; IR (KBr disc) (cm-1): 2985 (CH aliphatic), 1695 (C=O), 1570 (C=N); Anal. for C13H13N3OS2 (291.393) Calcd %, C, 53.58; H, 4.50; N, 14.42. Found C, 53.64; H, 4.47; N, 14.48; 1H-NMR (DMSO-d6): 0.9 (s, 3H,CH3) 1.29-2.55 (m, 10H cycloheptane); 13C-NMR (DMSO-d6) δ (ppm)=22.71-31.22 (6c), 117.40, 125.23, 139.56, 154.78, 155.31, 160.01, 164.36.
2-thioxo-6,7,8,9,10-pentahydrocyclohepta[4,5]thieno[2,3-d]-1,3,4-thiadiazolo[3,2-a]pyrimidin-11-one (10): Equivalent amounts (0.01 mol) of 7 and carbon disulfide were refluxed in pyridine (10 ml) for 8 h. After cooling, the reaction product was poured onto ice water and acidified with dilute hydrochloric acid, the formed solid was gathered after filtration using ethanol for crystallization to give thiadiazolothienopyrimidine derivative 10. Yield 69%, M.p. 166-168°C; IR (KBr disc) (cm-1): 3411 (NH), 3080 (CH aromatic), 2960 (CH aliphatic), 1700 (C=O), 1590 (C=N); Anal. for C12H11N3OS3 (309.43) Calcd%, C, 46.58; H, 3.85; N, 13.58; Found C, 46.66; H, 3.91; N, 13.61; 1H-NMR (DMSO-d6): 1.34- 2.97 (m, 10H cycloheptane), 10.84 (s, 1H, NH).
Biology
Cytotoxic screening on human Caucasian breast cancerous cells (MCF7)
The in-vitro anticancer screening method was performed in the Bioassay-Cell Culture Laboratory, National Research Centre, Dokki, Giza, Egypt. Viable cells were measured by reduction of the yellow MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) to the reduced purple formazan analogue [20]. The biological assay was performed in a sterile media using a Laminar flow cabinet biosafety class II level (Baker, SG403INT, Sanford, ME, USA). Cells were suspended in RPMI 1640 medium for MCF7. The media were enriched with 10% fetal bovine serum, 1% L-glutamine in addition to 1% antibacterial–antifungal mixture (10 000 U/ml potassium penicillin, 10 000 μg/ml streptomycin sulfate, and 25 μg/ml amphotericin B), and incubated at 37°C under 5% CO2. Cells were batch cultured for 10 days, then seeded at concentration of 10 x 103 cells/well in 96-well microtiter plastic plates at 37°C for 24 h in fresh complete growth medium under 5% CO2 using a water jacketed carbon dioxide incubator (Sheldon, TC2323, Cornelius, OR,USA). Media was suctioned, a medium without serum was introduced, and incubation of cells was carried out either having samples with various concentrations added or even alone (negative control) to result an overall concentration of (100–50–25–12.5–6.25–3.125–0.78 and 1.56 μM). Incubation was done for 2 days (48 h) then, medium was aspirated, 40-μl MTT salt (2.5 μg/ml) was introduced per well and further incubation was performed at 37°C for 4 h under 5% carbon dioxide. At 37°C overnight incubation the reaction was stopped and the formed crystals were dissolved upon addition of 200 μl of 10% sodium dodecyl sulfate (SDS) in deionized water per well [21].
A microplate multi-well reader (Bio-Rad Laboratories Inc., model 3350, Hercules, California, USA) was used to measure absorbance at 595nm using a reference wavelength of 620 nm. SPSS 11 program was used to test significance between samples and negative control (cells with vehicle) using independent t-test. DMSO was used as solvent with final concentration on the cells less than 0.2%. The percentage of change in viability was calculated according to the formula:
((samples result/negative control result) -1) x 100
Free radical scavenging activity
The in-vitro antioxidant activity method was performed in the Bioassay-Cell Culture Laboratory, National Research Centre, Dokki, Giza, Egypt. The free radical scavenging activity of the compounds was measured by 1,1-diphenyl-2-picryl-hydrazil (DPPH•) [22]. 0.1 mM of DPPH• was solubilized in methyl alcohol. At this point, 1 ml of this prepared alcoholic solution was added to 3 ml of the dissolved new thienopyrimidine tested samples at different concentrations ranging from 25 μM to 100 μM. The blend was shaken energetically and kept for half an hour at room temperature. Afterwards the absorbance was measured at λmax 517 nm in ASYS microplate reader taking into consideration that the minimum absorbance reflects maximum antioxidant effect.
The IC50 value for the tested compounds was measured statistically using nonlinear regression curve of Log concentration of the test compound (μM) against the mean percentage of the free radical scavenging effect.
DPPH scavenging effect (%) = 100−[ ((Z0-Z1)/Z0)× 100]
Z0 = absorbance without sample (control)
Z1 = absorbance with tested sample added [23]
The in-vitro antioxidant activity of the selected synthesized products was performed using the DPPH free radical scavenging method using Ascorbic acid as a reference standard. All compounds were screened at 100 μM and the % scavenging activity was illustrated in (Table 1, chart 1) as well as the IC50 and the IC90 of the most active compounds.
| Compound No. | IC50 (µM) | IC90 (µM) | % free radical scavenging at 100 µM |
|---|---|---|---|
| 3a | 57.4 | 101.9 | 85.2 |
| 3c | 63.8 | 107.9 | 70.7 |
| 4b | ----------- | ----------- | 52.7 |
| 4c | 34.6 | 56.9 | 100 |
| 5a | 21.1 | 46.2 | 100 |
| 6b | 18.1 | 42.8 | 100 |
| Ascorbic acid | 59.2 | 115 | 75.7 |
Table 1: IC50, IC90 and % free radical scavenging at 100 μM
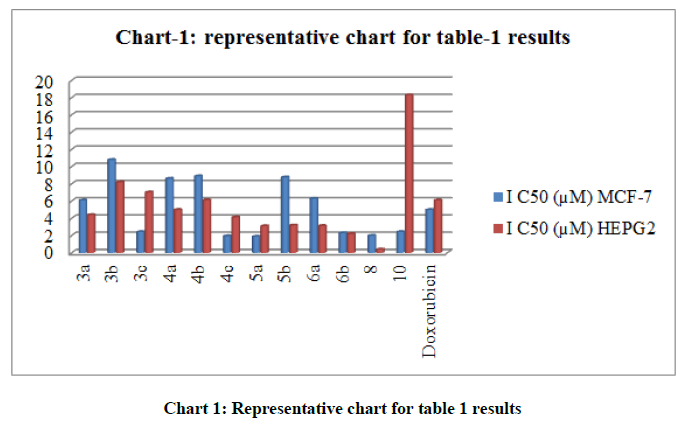
Chart 1: Representative chart for table 1 results
Results and Discussions
Chemistry
The plan strategy for syntheses of thienopyrimidines 1-10 is depicted in the provided (Scheme 1). The substituted thiophene ethylcarboxylate ester 1 [17] was prepared following reported Gewald reaction [18] using ethyl cyanoacetate, sulfur, cycloheptanone, and morpholine. The isothiocyanate derivative 2 is considered as a strategic starting material that can be used to synthesize a number of thieno[2,3-d]pyrimidines. It was prepared via the reaction of the amino ester 1 with thiophosgene as reported [19]. Interaction of isothiocyanate derivative 2 with different sulfonamides namely sulfanilamide, sulfadiazine and sulfamerazine were carried out in Dioxane to afford compounds 3a-c. IR spectra declared bands at 3350-3500 cm-1 representing NH. Moreover, 1H-NMR spectrum of 3a afforded multiplet at δ=7.12-7.49 ppm assigned for aromatic protons, in addition to three signals at δ=6.74, δ=10.83 and δ=11.50 ppm for the exchangeable protons NH2, NH and NH respectively. Furthermore, 13C-NMR spectrum of 3b showed signals attributed to aromatic-ring carbons at δ=126.2-138.2 ppm and signals at δ=110.31, 158.34, 169.30 ppm equivalent to pyrimidine carbons. Compounds 4a-c was obtained via the reaction of compounds 3a-c with hydrazine hydrate in ethanol under reflux. IR spectrum showed NH2 bands at ~3410 cm-1 in addition to two bands at 1338-1152 cm-1 attributed to SO2. 1H-NMR spectrum of 4b disclosed three interchangeable singlet signals at δ=4.11, δ=4.78, δ=11.65 ppm attributed to NH2, and two NH protons respectively. Signals assigned for pyrimidine protons at δ=6.90-7.40 ppm. For compound 4c, methyl protons were present at δ=2.35 ppm. 13C-NMR spectrum for 4b revealed signals for 21 carbons ranging from δ=25.76 ppm to δ=169.44 ppm.
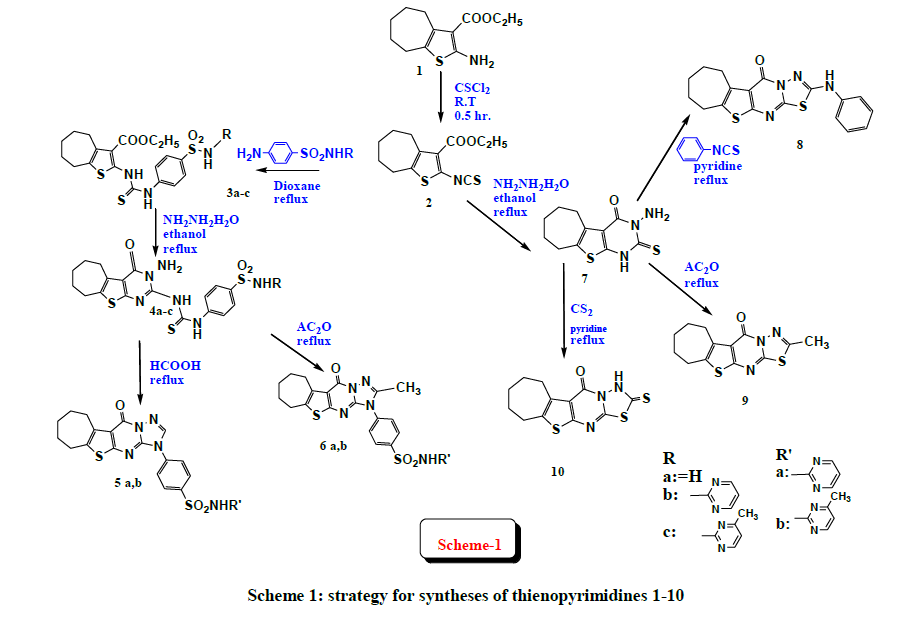
Scheme 1: strategy for syntheses of thienopyrimidines 1-10
Cyclization of compounds 4b and 4c into the corresponding triazolobenzenesulfonamide derivatives 5a and 5b respectively was done by reflux in formic acid. IR spectra confirmed the disappearance of the NH2 bands. 1H-NMR spectrum of 5a disclosed a singlet signal at δ=6.70 ppm representing the triazol proton, inaddition to multiplet signal at δ=7.30-7.87 ppm equivalent to aromatic-ring protons and the pyrimidine protons, furthermore a replaceable signal at δ=12.00 ppm assigned for NH proton. Moreover 13C-NMR spectrum for 5a revealed signal at δ=146.80 ppm representing triazol carbon and signals at δ=157.94, 158.20 and 169.61 ppm representing the pyrimidine carbons. Heating compounds 4b and 4c in acetic anhydride afforded compounds 6a and 6b respectively. IR spectra disclosed disappearance of the NH2 bands. 1H-NMR spectrum of compound 6a illustrated a singlet representing methyl protons of triazol ring at δ=2.33 ppm, multiplet at δ=7.20- 7.61 ppm assigned for protons of pyrimidine and aromatic ring, in addition to an interchangeable signal at δ=12.28 ppm correponding to NH proton.
Reflux of the isothiocyanate derivative 2 and hydrazine hydrate using ethanol as a solvent yielded 3-amino-2-thioxothienopyrimidinone derivative 7 upon acidification [24]. IR spectra confirmed NH2 bands at 3940 cm-1. 1H-NMR spectrum afforded two replaceable signals at δ=12.23 ppm and δ=13.21 ppm representing NH proton and NH2 protons respectively. In addition 13C-NMR spectrum showed signals at δ 161.40 ppm and δ=172.46 ppm corresponding to C=O and C=S respectively.
The reaction of 7 with phenyl isothiocyanate in refluxing pyridine was investigated yielded compound 8. IR spectra illustrated the presence of band for NH at 3390 cm-1. Moreover, 1H-NMR spectrum afforded multiplet signal at δ=7.16-7.49 ppm representing the aromatic-ring protons in addition to a replaceable signal at δ=12.23 equivalent for NH proton. Furthermore 13C-NMR revealed signal at δ=158.10 ppm attributed to carbon of thiadiazol ring in addition to signals representing aromatic carbons at δ=118.8-129.6 ppm.
The reaction of the 3-amino-2-thioxothienopyrimidinone compound 7 with acetic anhydride under reflux, afforded thiadiazolothienopyrimidine derivative 9. 1H-NMR spectrum afforded signal at δ=0.9 ppm assigned for methyl protons, moreover 13C-NMR spectrum afforded signal at δ=154.78 ppm equivalent to carbon of thiadiazol ring. Reaction of 7 with carbon disulfide was carried out in refluxing pyridine to afford thiadiazolothienopyrimidine derivative 10. IR spectrum confirmed the presence of band for NH at 3411 cm-1. In addition 1H-NMR spectrum disclosed an exchangeable signal at 10.84 ppm equivalent to NH proton.
Biological Discussion
The tabulated data in (Table 2, chart 2) demonstrate that the compounds with highest cytotoxic activity against MCF-7 reflected by their IC50S are: the triazolothienopyrimidine derivative 5a (IC50 1.90 μM) followed by the sulfamoylphenylaminothienopyrimidine derivative 4c (IC50 1.95 μM) then the thiadiazolothienopyrimidine derivative 8 (IC50 2.01 μM) compared to reference standard Doxorubicin (IC50 5.00 μM), while for those tested against hepatic cancer cell line HEPG-2, thiadiazolothienopyrimidine derivative 8 was the most potent (IC50 0.40 μM) followed by the multicyclic derivative 6b (IC50 2.21 μM) and 5a (IC50 3.11 μM) in comparison to Doxorubicin (IC50 6.10 μM). Moreover, an in-vitro antioxidant analysis was performed to measure the percentage of free scavenging activity for some of the best resulted anticancer compounds, where four out of six tested compounds elicited % of scavenging activity higher than that recorded by Ascorbic acid reference standard, which are compounds 4c, 5a and 6b that afforded 100 % scavenging and the thiouriedo derivative 3a that afforded 85.2 % compared to 75.7 % which was recorded by standard Ascorbic acid as shown in (Table 1).
| Compound No. | I C50 (µM) MCF-7 | I C50 (µM) HEPG2 |
|---|---|---|
| 3a | 6.09 | 4.4 |
| 3b | 10.8 | 8.19 |
| 3c | 2.44 | 7.03 |
| 4a | 8.62 | 5 |
| 4b | 8.9 | 6.12 |
| 4c | 1.95 | 4.14 |
| 5a | 1.9 | 3.11 |
| 5b | 8.77 | 3.2 |
| 6a | 6.3 | 3.14 |
| 6b | 2.31 | 2.21 |
| 8 | 2.01 | 0.4 |
| 10 | 2.44 | 18.3 |
| Doxorubicin | 5 | 6.1 |
Table 2: IC50s for some synthesized compounds against breast adenoma MCF-7 and liver adenoma HEPG-2 cell lines
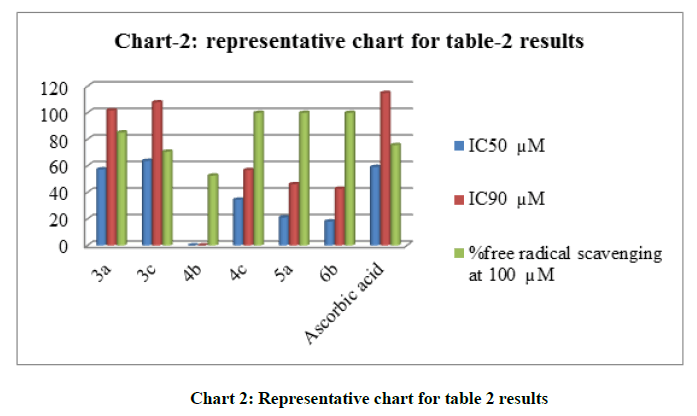
Chart 2: Representative chart for table 2 results
Conclusion
The biological evaluation declared that all the screened compounds revealed promising activity either for anticancer or antioxidant assays. For MCF-7 cell line-anticancer IC50 values were (1.90 μM-10.8 μM), six compounds showed remarkable cytotoxic activity more than that of Doxorubicin reflected by their IC50s ranging from (1.90 μM-2.44 μM) compared to standard Doxorubicin IC50 values (5.00 μM). For HEPG-2 cell line- the cytotoxic IC50 values were (0.4 μM -18.30 μM), eight out of twelve compounds illustrated better cytotoxic activity than the reference standard Doxorubicin as shown in their IC50 ranges (0.4 μM-5.00 μM) when compared to standard Doxorubicin IC50 values (6.10 μM). Whereas, their antioxidant % free radical scavenging was ranging from 52.7% to 100%, interestingly four of the tested compounds showed better antioxidant effect when in reference to standard Ascorbic acid, three of which had 100% free radical scavenging activity while that of standard Ascorbic acid was (75.7%). Noticeably, the compound’s antioxidant values were nearly consistent with their anticancer profile.
References
- Y. Babu, P. Chand, M. Wu, P.L. Kotian, V.S. Kumar, T.H. Lin, Y. El-Kattan, A.K. Ghosh, Therapeutic furopyrimidines and thienopyrimidines, 2006.
- W.J. Mc Clellan, Y. Dai, C. Abad-Zapatero, D.H. Albert, J.J. Bouska, K.B. Glaser, T.J. Magoc, P.A. Marcotte, D.J. Osterling, K.D. Stewart, S.K. Davidsen, M.R. Michaelides, Bioorg. Med. Chem. Lett., 2011, 21(18), 5620-5624.
- Y. Dai, Y. Guo, R.R. Frey, Z. Ji, M.L. Curtin, A.A. Ahmed, D.H. Albert, L. Arnold, S.S. Arries, T. Barlozzari, J.L. Bauch, J.J. Bouska, P.F. Bousquet, G.A. Cunha, K.B. Glaser, J. Guo, J. Li, P.A. Marcotte, K.C. Marsh, M.D. Moskey, L.J. Pease, K.D. Stewart, V.S. Stoll, P. Tapang, N. Wishart, S.K. Davidsen, M.R. Michaelides, J. Med. Chem., 2005, 48(19), 6066-6083.
- R.W. Luke, P. Ballard, D. Buttar, L. Campbell, J. Curwen, S.C. Emery, A.M. Griffen, L. Hassall, B.R. Hayter, C.D. Jones, W. McCoull, M. Mellor, M.L. Swain, J.A. Tucker, Bioorg. Med. Chem. Lett., 2009, 19(23), 6670-6674.
- T. Horiuchi, J. Chiba, K. Uoto, T. Soga, Bioorg. Med. Chem. Lett., 2009, 19, 305-308.
- E.I. El-Mongy, M.A. Khedr, N.A. Taleb, H.M. Awad, J. Heterocyclic Chem., 2017, 54(2), 1084-1093.
- N. Tani, M. Rahnasto-Rilla, C. Wittekindt, K.A. Salminen, A. Ritvanen, R. Ollakka, J. Koskiranta, H. Raunio, R.O. Juvonen, Eur. J. Med. Chem., 2012, 47, 270-277.
- K.S. Kumar, S. Chamakuri, P. Vishweshwar, J. Iqbal, M. Pal, Tetrahedron Lett., 2010, 51, 3269-3273.
- D.R. Gorija, K.S. Kumar, K. Mukkanti, M. Pal, Beilstein J. Org. Chem., 2011, 7, 338-345
- K.M. Al-Taisan, H.M.A. Al-Hazimi, S.S. El-Shih, Molecules., 2010, 15, 3932-3957.
- P.R. Murray, E.J. Baron, J.H. Jorgensen, M.A. Pfaller, R.H. Yulken, Manual of clinical microbiology. A.S.M., Washington, DC USA, 2003, 1212.
- M.M. Ali, A.E. Mohmoud, A.E. Rashed, Asian J. Pharm. Clin. Res., 2011, 4(1), 118-122.
- P. Rashmi, G. Laxmivenkatesh, N.K. Hazra, Der Chemica Sinica, 2011, 2 (2), 165-171.
- C.Y. Leung, A.M. Langille, J. Mancuso, Y.S. Tsantrizos, Bioorg. Med. Chem. 2013, 21, 2229-2240.
- P. Monique, F. Marylene, L.D. Cuong, H. Francois, G. Chantel, N. Guy, Eur. J. Med. Chem., 1988, 23(5), 453-456.
- H. Barua, N. Bhagat, M.P. Toraskar, Int. J. Curr. Pharm. Res., 2017, 9(1), 118-125
- L. Huangyong, C. Changshui, X. Shengzhen, Xiufang. Cao, J. Chem., 2013, 6.
- K. Gewald, E. Schink, H. Brottcher, Chem. Ber., 1966, 99, 94.
- N.A. Hassan, J. Sulfur. Chem., 2006, 27(6), 605-615.
- T. Mosmann, J. Immunol. Methods., 1983, 16, 65(1-2), 55-63.
- M.I. Thabrew, R.D. Hughes, I.G. McFarlane, J. Pharm. Pharmacol., 1997, 49, 1132.
- K. Shimada, K. Fujikawa, K. Yahara, T. Nakamura, J. Agric. Food Chem., 1992, 40, 945-948.
- M. Oktay, I. Gülçin, I. Küfrevio, Lebensm. Wiss. Technol., 2003, 36, 263-271.
- M.R. Prasad, J. Chem. Res., 2007, 3, 133-135.



