Review Article - Der Pharma Chemica ( 2018) Volume 10, Issue 7
A Review on Synthetic Methods for Preparation of Praziquantel
Pranaya P Dhawle and Anita S Goswami Giri*
Department of Chemistry, Chemistry Research Laboratory, VPM’s B. N. Bandodkar College of Science, Chendani Bunder Road, Thane (W), MS, India 400601
- *Corresponding Author:
- Anita S Goswami Giri
Department of Chemistry
Chemistry Research Laboratory
VPM’s B. N. Bandodkar College of Science
Chendani Bunder Road, Thane (W), MS, India 400601
Abstract
Praziquantel is synthesized pyrazineisoquinoline derivative having wide anthelmintic activity, and commonly used for treating all kinds of schistosomiasis. Synthesis of praziquantel generally involved diversified steps like Imino-Diels alder reaction, condensation, Ugi reaction, Nacylation, substitution, cyclisation and acylation followed by crystallization. It demonstrated high efficacy, low toxicity, short course treatment easy to use and affordability. Hence, present review is discussed landmarks of praziquantel with its process of preparation and application.
Keywords
Pharmaceutical ingredient gabriel phthalimide, Isoquinoline, Resolution, Pyrazinoisoquinoline derivative
Introduction
Schistosomiasis (bilharzias) is a disease of poverty endemic in 78 developing countries which was caused by parasites trematode flatworms that belongs to schistosomes. The disease affects 243 million people globally, caused due to poor sanitation. There are two main types of schistosomiasis viz. intestinal and urogenital. The schistosoma haematobium causes the urogenital schistosomiasis which damages bladder, uterus, and kidney. The parasites like Schistosoma guineensis, Schistosoma intercalatum, Schistosoma mansoni, Schistosoma japonicum and Schistosoma mekongi etc. are responsible for the intestinal schistosomiasis which causes liver and spleen enlargement, hypertension of abdominal blood vessel and damage of intestines [1-3].
Schistosomiasis was controlled to the great extent by proper drug treatment, social awareness, health education and proper sanitary measures [4]. When a person exposed with fresh water containing schistomes, due to any recreational or occupational activities it leads to schistosomiasis. The late diagnosis and treatment may lead to anemia, high morbididty and stunting. Numerical cases were reported as fatal [5] that was treated with an anthelmintic drug Paziquantel [5,6]. Praziquantel is the active pharmaceutical ingredient which used for the treatment of all types of schistosomiasis with great success. According to WHO policy on supply and availability of Praziquantel at reasonable cost leads to control shistosomiasis all over the world [7] hence, it was declared Praziquantel as an essential drug.
The distribution of Schistosomiasis throughout the world has consequences on economic and public health. hence required proper action plan to control the schistosomiasis [8,9]. The use of the safe and efficacious drug praziquantel, control the morbidity caused by schistosomiasis. To date, there is no evidence of development of clinically relevant resistance. Still there are no alternative drugs to PZQ with the possible exception of oxamniquin, although perhaps PZQ analogues could be developed [10]. Hence, present review focused on the landmarks of praziquantel drug.
Diversified scheme used for the synthesis of poraziquantel
Priorly, the 2-Acyl -4-Oxo-Pyrazino-Isoquinoline derivatives synthesized and claimed by S. Jurgen, et al. It explains reaction of 4-Oxo- 1,2,3,6,7,11b-hexahydro-4H-pyrazino[2,1-a]isoquinoline with different acid chloride to form praziquantel and analogues compound. The reaction is carried out in chlorinated solvent like chloroform in presence of organic base triethylamine (Scheme 1) [11].
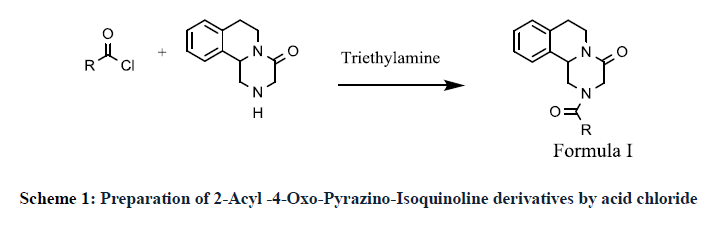
Scheme 1: Preparation of 2-Acyl -4-Oxo-Pyrazino-Isoquinoline derivatives by acid chloride
Berkowitz John, et al.were synthesized different heterocyclic tetrahydroisoquinoline, by internal imino-Diels Alder reaction with benzocyclobutenes and its derivatives (Scheme 2) [12]. The method of the tandem amidoalkylaion and N-acyliminium ion cyclization of amido-acetals was described by P. Joong H, et al. in the synthesis of pyrazinoisoquinoline derivatives including Praziquantel [13].
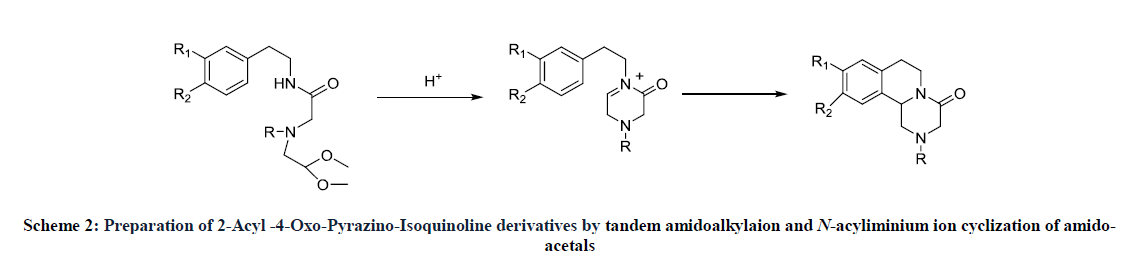
Scheme 2: Preparation of 2-Acyl -4-Oxo-Pyrazino-Isoquinoline derivatives by tandem amidoalkylaion and N-acyliminium ion cyclization of amido-acetals
The patent KR2002076486 provided synthesis of praziquantel by reacting Phenylethylamine with chloroacetylchloride to obtain 2-chloro-N-phenethylacetamide which then undergo Gabriel Phthalimide reaction to give corresponding amine 2-amino-N-phenylethylacetamide followed by treatment with bromoacetaldehyde dimethylacetal to give 2-[2,2(dimethoxy ethyl) amino]-N-(2-phenylethyl) acetamide. The above intermediate on further cyclization and acylation using cyclohexanoyl carbonyl produced praziquantel [14]. The invention in CN1683346, by S. Yuhua, L. Furongm et al. involves a similar process as mentioned in patent KR2002076486, except first step which involved 2- aminoacetylchloride reacted with phenylethyl amine which procured 2-amino-N-phenyl ethyl acetamide [15]. The PCT application WO2009115333 comprised novel process for the synthesis of praziquantel. In situ reaction, phenyl ethyl isonitrile, aminoacetaldehyde dimethyl acetal, formaldehyde and benzoic acid offered the intermediate which on further reaction converted into praziquantel (Scheme 3) [16].
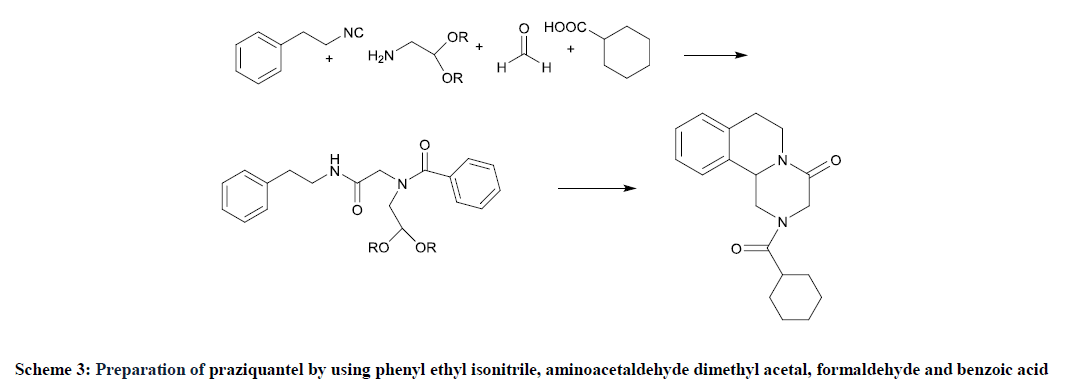
Scheme 3: Preparation of praziquantel by using phenyl ethyl isonitrile, aminoacetaldehyde dimethyl acetal, formaldehyde and benzoic acid
The synthesis of trioxaquantel derivatives as an innovative antischistosomal drugs involved process for synthesis of praziquantel and intermediates thereof [17]. The praziquanamine is synthesized by scheme,
Scheme 4 with four steps was claimed by S. Laurent J, et al. while five steps synthesis of praziquantel with low yield (16%) was reported by F. Yuste, et al. [18]. The compound N-(2, 2-dialkoxyethyl)(substituted-phenyl)alkylamines acts as intermediates in the preparation of heterocyclic compounds in the vein of tetrahydro–isoquinoline and its derivative thereof was described in US patent US3071613 [19]. The prevention of autoimmune diseases mediated by component or auxiliary of TNF-ot, NF-KB; IKK-ot, IKK-B, ATP-2 and p38 kinase by administration of praziquantel was stated in US 2006/0173011 [20].
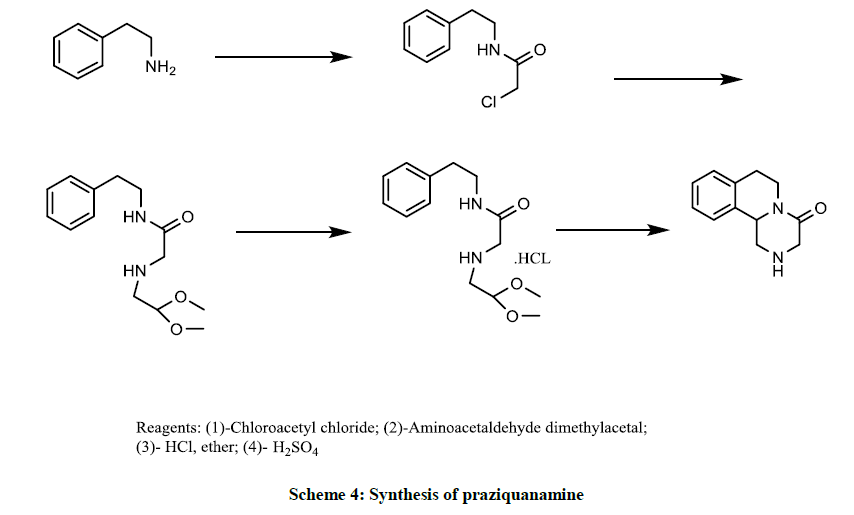
Scheme 4: Synthesis of praziquanamine
The analogues of the praziquantel were exhibited diversified biological activity was due to, arrival of divergent substituent on aromatic ring of praziquantel. The researcher gave depth idea about different amino substituted with aromatic rings by keeping remaining moiety was unchanged. These results have great importance in the choice of drug development for schistosomiasis shown in scheme 5 [21].
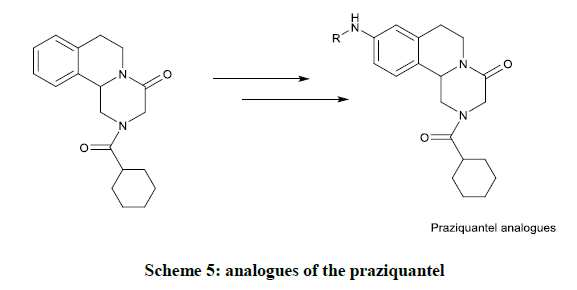
Scheme 5: analogues of the praziquantel
The Q. Chunhua, Chaomimng, et al. illustrated praziquantel derivatives containing aromatic ring which was substituted with -OH group on C-10. The –OH of praziquantel is efficacious against adolescent and adult worm of the schistosomiasis, whereas the praziquantel is effectual against adult worm indicating -OH group on C-10 of praziquantel is paramount substitute for the treatment of schistosomiasis than divergent praziquantel [22].
A single-step synthesis of 4-oxo-hexahydro-pyrazinoisoquinoline derivatives exhibited better yield comprising hydrolyzing a 2-acyl-4-oxo- 1,2,3,6,7,1 lb –hexahydro -4H-pyrazino-[2,1-a]isoquinoline with oxygen containing acid or acid salt of inorganic pentavalent phosphorus or hexavalent sulfur was claimed in US patent US 4049659. The reaction of 2-benzoyl-4-oxo-1,2,3,6,7,11b-hexahydro-4H-pyrazino[2,1- a]isoquinoline with 98% phosphoric acid for 2 days at 100° C offered 4-oxo-1,2,3,6,7,11b-hexahydro-4H-pyrazino[2,1-a]isoquinoline.
Synthesis of 2-(3,4,5-trimethoxybenzoyl)-4-oxo-6-trans-methyl-1,2,3,6,7,11b-hexahydro-4H-pyrazino[2,1-a] isoquinoline from 1-cyano-2- (3,4,5-trimethoxybenzoyl)-1,2-dihydroisoquinoline by hydrogenation, followed by treatment with chloroacetyl chloride and ring closure of the resultant 1-[(3,4,5-trimethoxybenzoyl)-aminomethyl]-2-chloroacetyl-1,2,3,4-tetrahydro isoquinoline with potassium tert.-butylate and further treatment with 98% phosphoric acid to give claimed compound. Furthermore, it comprised processes for preparation of different derivatives like (-)-2-benzoyl-4-oxo-1,2,3,6,7,11b-hexahydro-4H-pyrazino[2,1-a]isoquinoline and 4-oxo-1,2,3,6,7,11b-hexahydro-4H-pyrazine[2,1- a]isoquinoline, etc. [23].
The anthelmintic resinates and a method for their preparation was developed by L. Hughes, et al. in US patent US 2004/0180034 A1, which provided a pharmaceutical composition that consisting of a basic anthlemintic drugs such as Praziquantel, Epsiprantel, Droncit, etc. [24].
The multi-step synthesis of 2-Palmitoyl [1,2,3,6,7,11b]hexahydro-4H-pyrazino[2-la]isoquinoline-4-one a lipophilic, analogue of the Praziquantel which is compatible to the human metabolically and physiologically was put forwarded by A. Soliman, et al. [25].
The L. Balaya, et al. specified a novel and cost-effective process for the synthesis of Praziquantel by introducing novel intermediate, 2-[(2,2- dimethoxyethyl)benzyl amino]-N-phenethylacetamide. The synthesized Praziquantel was in novel crystalline form [26]. J. Seubert and H. Thomas discussed in the US patent US.4,866,056 preparation of praziquantel from Benzazepine [27], whereas one step reaction by condensation between 2-piperazinone with substituted benzoic acids which was substituted 4-(2-, 3- and 4-R)-carboxyphenyl-1,4-pyrazin-2-ones interpreted by R. Martínez, et al. [28].
Praziquantel synthesis steps involved (i) hydrolysis of 1, 2, 3, 6, 7, 11beta-hexahydro-4H-pyrazino[2,1-alpha]isoquinolin-4-one and phosphoric acid, distillation under vacuum, (ii) addition of deionized water for dilution and crystallization to observred a phosphate compound intermediate; (iii) acylation using cyclohexaformyl chloride in an organic solvent in the presence of base to obtain a praziquantel crude product; (iv) decolorisation and recrystallisation of crude gives pure praziquantel [29].
One-pot method for preparing praziquantel, involved steps: (i) reaction of chloroacetaldehyde dimethyl acetal and an ammonia aqueous solution to generate aminoacetaldehyde dimethyl acetal; (ii) a condensation reaction of beta-phenylethylamine and chloroacetyl chloride to generate an intermediate 1; (iii) condensation of the intermediate 1 and the aminoacetaldehyde dimethyl acetal to generate an intermediate 2; (iv) cyclization of the intermediate 2 in the presence of an acidic catalyst to obtain an intermediate 3; (v) acylation of the intermediate 3 using cyclohexanecarbonyl acid chloride in an organic solvent under basic condition to offer praziquantel [30].
Y. Chuanxin, et al. learned the structural elucidation of formula I (Scheme 1), which was effective as an anti-schistosomiasis drugs along with active for polypide paralysis, and insecticide. It overcomes less curative effect or ineffective treatment or drug resistance caused due to long-term use of the anti-schistosomiasis drugs in the prior art [31].
Praziquantel was also synthesized using glycine ethyl ester and cyclohexanecarboxylic acid chloride as starting materials, which reacts with bromoacetoaldehyde dimethyl acetal and beta-phenylethylamine. The final step was an annulation reaction which offers praziquantel. The method incorporated with cheap and easily available raw materials, mild reaction conditions and low toxicity [32]. The patent CN105294679 (A) by L. Jingpeng, Z. et al.disclosed high-purity praziquantel from cheap and readily -available raw materials via safe and simple reactions under mild reaction conditions [33].
The praziquantel synthetic technology included beta-phenylethylamine, amino acetyl halide hydrochloride, halogenated acetaldehyde alcohol and cyclohexyl formyl chloride as raw materials, which undergo series of reaction like condensation, cyclization and acylation which results into praziquantel. The synthetic technology has the advantages of mild reaction conditions, room temperature and atmospheric pressure operation. Overall yield of the process is more than 50% [34].
The patent CN105622606 (A) by Y. Shunfu, et al. related for the synthesis of praziquantel involving is simple and safe steps (i) reaction on phenylethylamine with chloracetyl chloride; (ii) addition of the steps (i) into a hydrogen debenzylation solution to react steps (i) product (iii) steps (ii) product was decolorized; and (iv) cyclization with cyclohexyl formyl chloride in an alkaline medium procured crude praziquantel [35].
Recently in 2018, US patent US2018030049 (A1) prepared antihistaminic drug praziquantel and intermediates by reacting β-phenethylamine with chloroacetyl chloride, and subsequent substitution reaction with ethanolamine, and an acylation reaction with cyclohexanecarbonyl chloride, followed by an oxidation and cyclization. It also claim two key intermediates in the synthesis of praziquantel, specifically a compound of formula IV and a compound of formula V (Scheme 6) [36].
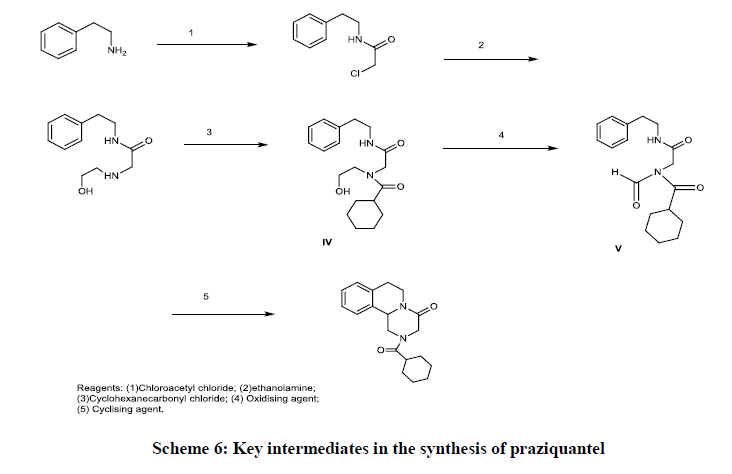
Scheme 6: Key intermediates in the synthesis of praziquantel
Resolution of Praziquantel
The method for the racemization of enantiomerically pure praziquantel under basic conditions and the process for the preparation of optically pure form of (R)- praziquantel was stated in patent CN107108607 (A) [37].
The simple, environmentally safe, one-pot synthesis was used in the production of praziquantel with the course of reaction containing acylation of beta-phenylethylamine by using chloroacetyl chloride and liquid caustic soda in dichloromethane as a solvent [38].
The optically pure (R)- praziquantel and its analogues with method of preparation was best explained by E. Luc, et al. The reactions proceed are stereoselective. It includes process which involves asymmetric hydrogenation of the intermediate compound (I) and its subsequent cyclization (Scheme 7) [39].
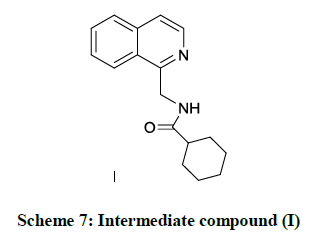
Scheme 7: Intermediate compound (I)
The procedure for production of 4-benzyl-1-phenethyl-piperazin-2,6-dione, intermediate III and intermediate IV involved the process for synthesis of intermediate IV (Figure 1), which is used as key intermediate in the synthesis of praziquantel [40].
The racemic praziquanamine-1 and recovery of isomer praziquanamine S-1 conversed to the racemic compound 1. The resolved compound was L-praziquantel [41].
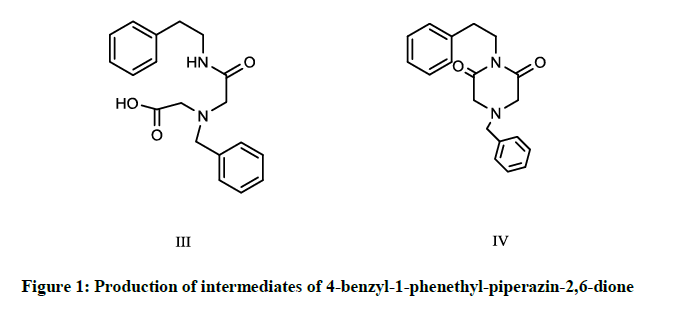
Figure 1: Production of intermediates of 4-benzyl-1-phenethyl-piperazin-2,6-dione
The Woelfle, et al. mentioned two methods for resolution of praziquantel as a single enantiomer. Initial method involves a hydrolysis of commercially available praziquantel to an intermediate amine, which is resolved with a derivative of tartaric acid. The subsequent method employs a different intermediate that may be resolved with tartaric acid itself [42].
P. Roszkowski, et al. made available the process for preparation of enantiopure form of Praziquantel via asymmetric transfer hydrogenation according to the Noyori protocol (Scheme 8). The reduction of prochiral imine 4 affords product 5 in 62% ee, but a single crystallization leads enantiomeric purity to 98% ee [43].
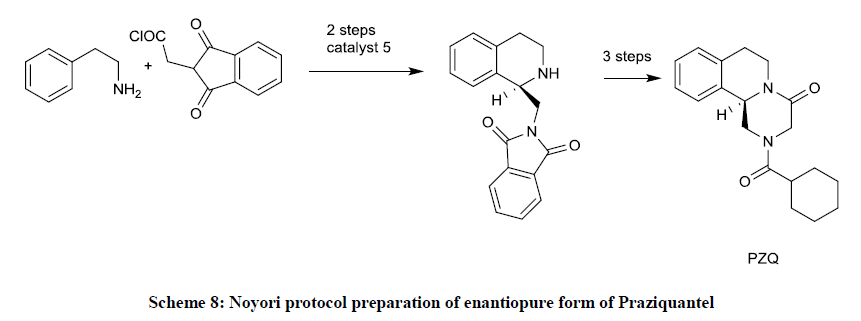
Scheme 8: Noyori protocol preparation of enantiopure form of Praziquantel
Preparation of optically pure levo praziquantel and its an intermediate thereof involved salt forming reaction on a compound of formula (I) (Figure 2) and its resolution using alkaline resolving agent gave levo praziquantel intermediate of formula (II) (Figure 2); which then undergo series of reaction to synthesize levo praziquantel [44].
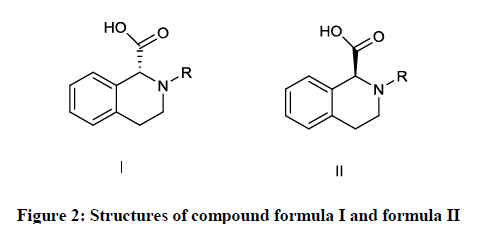
Figure 2: Structures of compound formula I and formula II
The Method for preparing (R)-praziquantel was described by Qian, M. [45]. That related to a novel method for preparation of (R)-praziquantel by fascinating benefit of the high stereo selectivity, site selectivity and region selectivity of an enzyme. It results into optically pure, chiral (R)- praziquantel which was achieved by means of the dynamic kinetic resolution of an enantiomer from the synthesized racemate or derivatives thereof. The (R)-praziquantel was obtained from conventional and mature organic chemical reactions with higher yield [45].
The crystal form of (r)-praziquantel was superior to existing crystal form of praziquantel with better solubility, better drug efficacy, better pharmacokinetic characteristics, reproducible results, environmentally friendly, low cost, able to operate at a normal pressure and temperature, and also suitable for large-scale production. The characterization of R-Praziquantel involves X-ray diffraction pattern (CuKα radiation) at 25°C. It results the following diffraction peaks: 2-Theta=6.9 ± 0.2°, 8.3 ± 0.2°, 15.1 ± 0.2°, 17.4 ± 0.2°, 19.8 ± 0.2°, 21.9 ± 0.2°, 24.3 ± 0.2° or d=12.74 ± 0.20 Å, 10.61 ± 0.20 Å, 5.87 ± 0.20 Å, 5.09 ± 0.20 Å, 4.48 ± 0.20 Å, 4.06± 0.20 Å, 3.66 ± 0.20 Å [46].
The new intermediates for the synthesis of (R)-praziquantel and process for preparation involved - condensation, reduction and acylation followed by ring-closing reaction. It involves use of (1R)-2-substituted-1,2,3,4-tetrahydroisoquinoline-1-carboxylic acid as starting material. The said process provides optically pure (R)-praziquantel product, overall process is cost effective [47].
The Q. Mingxin claimed in CN107151246 (A), the preparation method of (R)-praziquantel amine salt, and a method of synthesis of levopraziquantel. The reaction of racemic praziquantel amine and a particular chiral resolution reagent in a solvent offers(R)- praziquantel amine salt. The chiral resolution reagents claimed were (R)-ibuprofen, (R)-naproxen and (R)-phenethylsulfonic acid. The methods have the advantages like readily available and inexpensive raw materials, simple process, short duration and affordability of process. It was also associated with recovery of chiral resolution reagent [48]. The synthesis of series of tetrahydroisoquinoline compounds containing halide (specifically Bromo-) substituent on aromatic ring is innovated (by Z. Wang, et al.).
Derivatives of praziquantel
The current embodiment involved preparation of modified praziquantel derivatives and its evaluation against juvenile and adult stage of S. japonicumin. Most of the isolated analogs showed equivalent activity to the praziquantel. The in vitro and in vivo activity against worm’s japonuicum schistosomesis also deliberated. The compound with bromo substituent on the aromatic ring of praziquantel revealed close anti-schistosomal activity to parent drug praziquantel [49].
The Deuterated and Fluoroderivatives of pyrazinoisoquinoline were patented in US US8889687B2. This invention gives novel compounds that were pyrazinoisoquinoline derivatives and their pharmaceutically acceptable salts. It encompassed of novel pyrazinoisoquinoline derivatives which were derivatives of praziquantel (Figure 3) such as Dueterated derivatives, Fluoroderivatives etc. The process of synthesis of praziquantel incorporated the use of Aminoacetaldehydedimethylacetal [50].
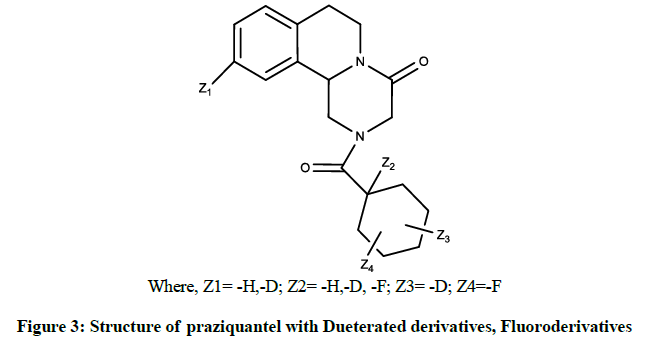
Where, Z1= -H,-D; Z2= -H,-D, -F; Z3= -D; Z4=-F
Figure 3: Structure of praziquantel with Dueterated derivatives, Fluoroderivatives
The Deuterated pyrazinoisoquinoline (Figure 4) compound with R-configuration [51] is as follows-
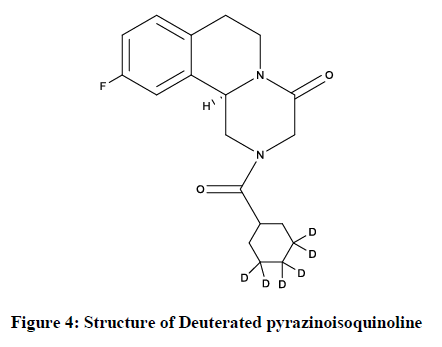
Figure 4: Structure of Deuterated pyrazinoisoquinoline
Thirty novel PZQ derivatives were described based on the Ugi 4-component reaction discussed by Haixia Liu et al. All derivatives were slightly less active than the mother drug PZQ [52]. A simple and sensitive method for the polarographic determination of praziquantel (1) after derivatization using Vilsmeier formylation is described [53]. Solid phase synthesis was also used for synthesis of praziquantel [54]. Synthesized praziquantel amide and urea derivatives had low to modest activity against juvenile worms while ketone derivative did not exhibited any activity. But combination therapy was worked against schistosomiasis [55]. The synthesized ferrocenyl derivatives of the drug praziquantel demonstrated anthelmentic activity in the micromolar range which was not sufficient for in vivo testing [56]. A structural variation in praziquantel analogues at piperazine, amide, and aromatic moieties influenced the modest to low activity against adult S. mansoni [57]. Cellulose derivative of praziquantel with chiral stationary phases was prepared with spacer reagent, bifunctional reagent of 3-(triethoxysilyl) propyl isocyanate which affects the resolution [58].
The reaction of praziquantel and furoxan moieties in a single entity produces hybrid compounds called NO-donor praziquantel exhibited biological activity. All the derivatives show activity to inhibit recombinant S. mansoni thioredoxin glutathione reductase (TGR) [59]. Wang, et al. described synthesis and the structure-activity relationship studies of newly synthesized praziquantel derivatives. Most of the derivatives showed better biological activity against S. japonicum than praziquantel whereas some of them were less effective for treatment of schistosomiasis [60]. Number of researchers synthesized and analysed praziquantel and derivatives with structural modification they show inhibition of Schistosomiasis, with less or more efficacy.
Conclusion
The praziquantel is well known for its anthelmintic activity. Modification in aromatic ring at C-10 with hydroxyl group of praziquantel showed multi fascinated activity than the praziquantel. Researcher used single step, two steps and multi steps reactions for the synthesis of praziquantel. Many intermediates and there derivatives are ecofriendly, easily available, having high efficacy hence used to treat infectious diseases such as all types of schistomes and liver flukes. The review reported mode of synthesis and resolution with scope of modification in praziquantel synthesis. Because mentioned methodologies are associated with disadvantages like strong reagents used in synthesis, harsh reaction condition, expensive raw materials, excess use of organic and halogenated solvent etc., as praziquantel is broad spectrum essential drug, it is crucial to develop simple, cost effective, green and time saving method to synthesize praziquantel.
References
- E. Pearce, A. MacDonald, Nature Rev. Immunol., 2002, 2(7), 499.
- B. Gryseels, Human schistosomiasis, The Lancet, 2006, 368(9541), 1106-1118.
- X. Zhou, Acta Tropica., 2005, 96(2-3), 97-105.
- S. Luiz, J. Marcal, C. Glasser, Control of Schistosomiasis Transmiss., 1995, 285-288, 90(2).
- A. Domling, K. Khoury, Praziquantel and schistosomiasis. Chem. Med. Chem., 2010, 1420-1434.
- D. Cioli, L. Pica-Mattoccia, Parasitol. Res., 2003, 90(1), S3-S9.
- P. Andrews, Med. Res. Rev., 1983, 3(2), 147-200.
- L. Chitsulo, D. Engels, A. Montresor, L. Savioli, Acta tropica., 2000, 77(1), 41-51.
- A. Fenwick, L. Savioli, D. Engels, N. Robert, M. Todd, Trends in parasitology., 2003, 19(11), 509-515.
- A. Domling, K. Khoury, Chem. Med. Chem., 2010, 5(9), 1420-1434.
- H. Kim, S. Yong, H. Park, C. Kim, Tetrahedron., 1998, 54(26), 7395-7400S. 1973.
- Jurgen, T. Herbert, A. Peter, 2-Acyl-4-oxo-pyrazinoisoquinoline derivatives and process for the preparation thereof, U. S. Patent 4001411, December 17.
- W. Berkowitz, T. John, The J. Org. Chem., 1984, 49(26), 5269-5271
- KR2002076486, Google Patents.
- S. Yuhua, L. Furong, Praziquantel synthetic process, CN1683346, March 1,2005.
- A. Doemling, Novel synthesis of praziquantel,WO2009115333, September 24, 2009.
- S. Laurent, J. Boissier, F. Cosledan, Eur. J. Org. Chem., 2008, 895-913.
- F. Yuste, Y. Pallas, J. Heterocyc. Chem., 1986, 189-190.
- A. Surrey, N-(2, 2-dialkoxyethyl)-(halogenated-phenyl) alkylamines, US3071613A, July 03, 1957.
- D. Shin, T. Choi, M. Cho, Treatment of inflammatory disorders with praziquantel, US2006173011 (A1), Jan 18, 2005.
- F. Ronketti, A. Venkata Ramana, X. Chao-Ming, M. Todd, Bioorg. Med. Chem. Lett., 2007, 17(15), 4154-4157.
- C. Qiao, C. Xia, Preparation method of 10-hydroxyl praziquantel and application thereof as anti-schistosomiasis medicine, CN102285985B, August 08, 2012.
- P. Rolf, Process for the preparation of 4-oxo-hexahydro-pyrazinoisoquinoline derivatives,US 4049659, September 20, 1977.
- L. Hughes, W. Ziarno, Anthelmintic resinates and a method for their preparation, EP1457213A1, March 10, 2003.
- M. Abo-Ghalia, A. Soliman, Acta poloniae pharmaceutica, 1999. 57(1), 53-59.
- B. Lingappa, M. Mahalinga, D. Yogeesh, P. Kakrannaya, Process for the preparation of praziquantel, US 8,754,215, June, 17, 2014.
- R. Dorgan, R. Elliott, Benzazepines and methods therefor, US4866056A, July 18, 1983.
- R. Martínez, M. Rubio, R. Toscano, J. Heterocyc. Chem., 1994, 31(6), 1521-1523.
- Y. Congyong, L. Jinbiao, Z. Tao,Praziquantel preparation process, CN103059018 (A), April 24, 2013.
- X. Lliuhua, L. Mingming, X. Jiangjun, L. Jiagen, Method for preparing praziquantel, CN103739601, April 23, 2014.
- Y. Chuanxin, F. Bainian, S. Lijun, W. Wenlong, Praziquantel analogue, preparation method and application thereof, CN103910725, July 9, 2014.
- S. Miaogen, X. Xaoli, Novel preparation method for praziquantel, CN105153157, December 16, 2015.
- L. Jingpeng, Z. Nan, L. Guangshun, Preparation method for praziquantel, CN105294679, February 03, 2016.
- Z. Jianying, Praziquantel synthetic technology, CN105622604, June 01, 2016.
- Y. Shunfu, Y. Jun, Y. Zhiping, Preparation method of praziquantel, CN105622606, June 01, 2016.
- Z. Fuli, Y. Zhezhou, B. Rusheng, X. Wiwei, Preparation method for praziquantel and intermediate compounds thereof, U. S. Patent 2018030049, February 1, 2018.
- W. Andreas, S. Hadia, Christian,; Joern, K.; Method for the production of praziquantel. CN107108607, August 29,2017.
- W. Feng, F. Jun, L. Jiasheng, W. Zikun, A process of synthesizing praziquantel, CN106866663, June 20, 2017.
- E. Luc, W. Andreas, M. David, L. Stefan, Method for the production of praziquantel and precursors thereof, WO2017036577, March 09, 2017.
- Method for production of 4-benzyl-1-phenethyl-piperazin-2,6-dione, intermediate compound and method for its production. RU2015152291 (A), July 28, 2017.
- Z. Fuli, Y. Zhezhou, Racemic praziquanamine recovery and preparation methodCN107151244 (A), September 12, 2017.
- M. Woelfle, J. Seerden, J. Gooijer, K. Pouwer, P. Olliaro, PLoS Negl. Trop. Dis., 2011, 5(9), e1260.
- P. Roszkowski, J. Maurin, Z. Czarnocki, Tetrahedron: Asym., 2006, 17(9), 1415-1419.
- L. Wensen, Y. Wansong, Preparation method of levo praziquantel as well as intermediate thereof, CN103539796, May 20, 2015.
- M. Qian, Method for preparing (R)-praziquantel, U. S. Patent 9068214, June 30, 2015.
- Q. Mingxin, H. Rodney, Crystal form of (r)-praziquantel and preparation method and application thereof, U. S. patent 20160272636, September 22, 2016.
- Q. Mingxin, Process and intermediates for the synthesis of (r)-praziquantel, U. S. Patent 2017121330,October 31, 2017.
- Q. Mingxin, Preparation method of (R)-praziquantel amine salt, and preparation method of levopraziquantel, CN107151246, September 12, 2017.
- Z. Wang, J. Chen, C. Qiao, Chem. Biol. Drug design., 2013, 82(2), 216-225.
- S. Harbeson, R. Tung, Pyrazinoisoquinoline compounds, U. S. Patent 8889687B2, November 18, 2014.
- J. Liu, R. Tung, S. Harbeson, Deuterated pyrazinoisoquinoline compounds, U. S. Patent, 8, 889, 687, November 18, 2014.
- H. Liu, S. William, E. Herdtweck, S. Botros, A. Dömling, Chem. Biol. drug design., 2012, 79, 470-477.
- E.J. Kim, W. Hänsel, D. Heber, Pharmazie., 2001, 56(2), 146-149.
- S. El-Fayyoumy, W. Mansour, M.H. Todd, Tetrahedr. Lett., 2006, 47, 8, 1287-1290.
- D. Yuxiang, C. Jacques, V. Mireille, R.M. Nuha, B. Quentin, A. Yazen, H. Jiangeng, K. Jennifer, V. L. Jonathan, Bioorg. Med. Chem. Lett., 2010, 20(8), 2481-2484.
- M. Patra, K. Ingram, V. Pierroz, S. Ferrari, B. Spingler, J. Med. Chem., 2012, 55(20), 8790-8798.
- P. Sadhu, S. Kumar, M. Chandrasekharam, L. Pica-Mattoccia, Bioorg. Med. Chem. Lett., 2012, 22, 1103-1106.
- X. Chen, Y. Liu, F. Qin, Li, J. Chromatogr. A., 2003, 1010, 185-194.
- S. Guglielmo, D. Vottero, D. Williams, R. Fruttero, A. Gasco, Eur. J. Med. Chem., 2014, 84, 135-145.
- W. Wang, L. Song, X. Chen, X. Yin, G. Wang, Molecules., 2013, 18(8), 9163-9178.



