Review Article - Der Pharma Chemica ( 2018) Volume 10, Issue 6
A Review on Antibacterial Activity of Some Isoindole Derivatives
Ferenc Csende* and Andrea Porkoláb
Taxus Research Laboratory, Bocskai u. 22., H-4080 Hajdúnánás, Hungary
- *Corresponding Author:
- Ferenc Csende
Taxus Research Laboratory
Bocskai u. 22., H-4080 Hajdúnánás, Hungary
Abstract
Compounds which are having an isoindole moiety are important classes of the bioactive molecules. Several isoindoles and their fused derivatives were synthesized and evaluated for their efficiency as antimicrobial agents in the past years. However, several novel problems appeared with the treatment of various infections (resistant bacterial or nosocomial infections, multidrug-resistant bacterial, rare and aggressive infections), thus medicinal or pharmaceutical chemists are challenged to discover and find improved novel and more efficient drugs. The aim of this overview is to summarize the most efficient and promising isoindole derivatives to promote these developments of potent and safe drugs with fewer side effects.
Keywords
Antibacterial, Enzymes, Inhibition, Isoindoles, Fused rings
Introduction
The synthesis, chemistry, structural and pharmacological behaviour of isoindoles were earlier surveyed thoroughly [1]. Isoindoles, constitutional isomers of indoles and their derivatives are less well studied. The isoindole shows structural characteristics indicative of an aromatic system: the carbocyclic ring shows bond alternation reminiscent of that found in naphthalene [2]. Isoindole is much less stable than its isomer indole, thus the saturated, fused and 1- or 1,3-dioxo derivatives are used in the organic and pharmaceutical chemistry practically [3]. The isoindoles are important groups of N-heterocyclic compounds occur widely in living matter and have significant biological roles [4]. Isoindoles constitute the core of many alkaloids, phthalocyanines, synthetic fused heterocycles, isoindoline pigments and isoindole-BODIPY dyes [5-7]. In this overview we could report some isoindole derivatives with regard to their antimicrobial activity. Although most bacteria will not invade other living organism and many more bacteria are harmless to our immune systems or often beneficial, some are pathogenic, with the number of species estimated as fewer than hundred that are seen to cause infectious diseases in humans. People can get infected with pathogenic bacteria by direct (physical) or indirect contacts (water, food, water, wounds and biological products like blood, serum, plasma and airborne transmission). The variety of pathogenic bacteria is quite broad, but there are some that commonly cause infectious diseases. The most common pathogenic bacteria are the Staphylococcus aureus, some Streptococcus species and certain strains of Escherchia coli, they cause also some of the most serious diseases. The cholera is caused by the bacterium Vibrio cholerae and usually spreads by drinking infected water and due to unhealthy conditions [8]. There are the main classes and types of the antibacterial drugs: penicillins, aminoglycosides, carbapenems, cephalosporins, glycopeptides, macrolides, polypeptides, quinolones, sulfonamides and finally tetracyclines. The development of new medicines becomes the important and urgent target because of the limited availability of efficient broad-spectrum antibiotic drugs and the increasing evolvement of resistance.
7-Amino- and chloro-isoindole derivatives
Neumann H et al., [9] repoted that some N-analogue of the corollosporine have notable antibiotic activity, the compunds of which were prepared by the conversion of N-methyl-4-aminophthalimides with different equivalents of Grignard reagents. Thus 7-amino-3-hexyl-3-hydroxy-2- methyl-2,3-dihydroisoindol-1-one and its 2,4,6-trimethyl derivative (Figure 1) were found to be active against Staphylococcus aureus (inhibition zone: 10 mm).
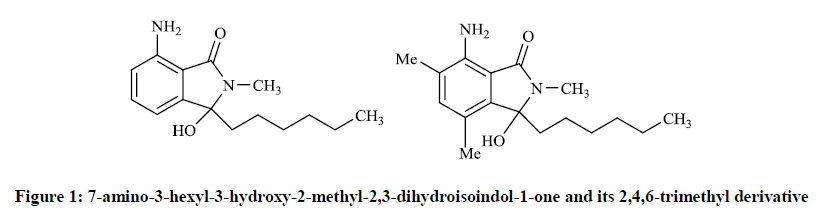
Figure 1: 7-amino-3-hexyl-3-hydroxy-2-methyl-2,3-dihydroisoindol-1-one and its 2,4,6-trimethyl derivative
Some of the chlorinated compounds (Figure 2) showed activity according to the screening results with the minimal inhibitory concentration (MIC) values of 83.5 and 28.5 μg/ml againts Bacillus subtilis.
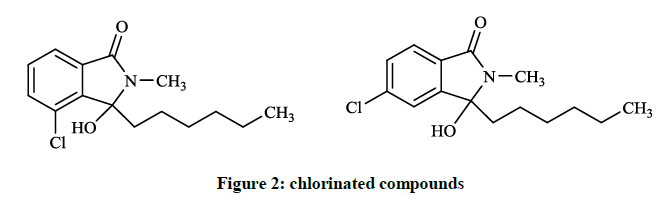
Figure 2: chlorinated compounds
Mikolasch A et al., [10] synthesized further N-analogous corollosporines (Figure 3) via laccase-catalyzed coupling reaction of the N-methyl-4- aminophthalimides with 2,5-dihydroxybenzoic acid derivatives. The obtained products (R = H, Me) were tested in several biological assay to evaluate their antimicrobial effect. According to the results, the compounds prepared inhibited the growth of several gram positive bacterial strains, among them methicillin-resistant S. aureus (inhibition zone: 8-10 mm).
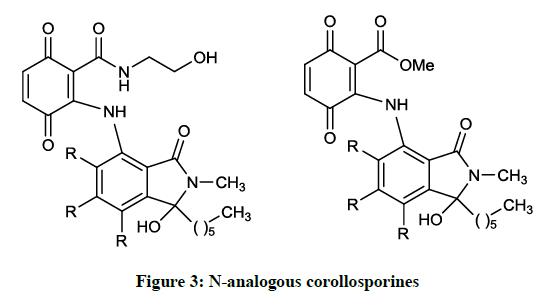
Figure 3: N-analogous corollosporines
Tricyclic β-lactams (trinems or tribactams)
The trinems (tribactams) are a novel class of antibiotics, which are tricyclic β-lactams containing a unsaturated or partly saturated isoindole moiety. Heck JV et al., [11] synthesized the fused analogue of the β-lactam Nocardicin A (Figure 4), which is more active against gram negative than gram positive microorganisms. The preparation of the tricyclic cyclonocardicin was achieved starting from 2-bromo-5- hydroxybenzaldehyde in a multi-step procedure. Unfortunately, the isoindole analogue had quite poor stability in neutral aqueous solution and the antibacterial activity was less effective. Christensen BG et al., [12] prepared 6-(1-hydroxyethyl)-cyclonocardicin (Figure 5) by a similar method. This compound is also a novel class of antibiotics, which is bioactive against a broad range of pathogens (e. g. S. aureus, E. coli, Klebsiella pneumoniae, B. subtilis).
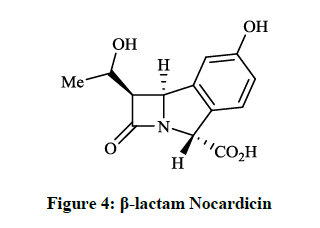
Figure 4: β-lactam Nocardicin
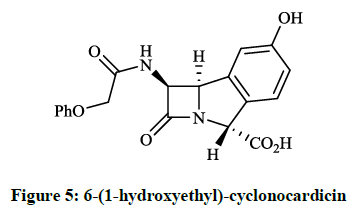
Figure 5: 6-(1-hydroxyethyl)-cyclonocardicin
Hammond SM et al., [13] investigated the in vitro activity of hexahydroazeto[2,1-a]isoindol-2(1H)-one namely, tribactam GV104326 (Figure 6) against gram positive, gram negative and anaerobic bacteria. This compound was extremely active against the Gram-positive bacteria tested, such as S. aureus strains and Staphylococcus epidermidis were highly sensitive to GV104326.
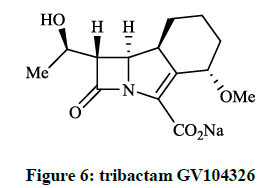
Figure 6: tribactam GV104326
The cummulative MICs were for 90% of the tested strains (MIC90s) ≤ 0.12 μg/ml and MIC90 ≤ 0.06 μg/ml respectivelly. Among other tested antibiotics the GV104326 exhibited consistently low MIC90s for S. aureus, E. coli, K. pneumoniae, Klebsiella oxytoca, Proteus mirabilis, Proteus vulgaris and Clostridium perfringens. Additional in vitro tests and in vivo microbiological evaluations of the tricyclic β-lactam GV104326 (Sanfetrinem as sodium salt and Sanfetrinem cilexetil ester, which is a prodrug) were carried out in detail [14-16]. The orally bioavailable and metabolically labile ester derivative (GV118819) is currently undergoing phase II clinical trials. Several methods were developed and patented for the synthesis of GV104326 and its derivatives [17-21]. Andreotti D et al., [22] described the synthesis of all the isomers of 8-methoxy- and 4-methoxy derivatives of compound GV104326 and evaluated their in vitro antibacterial activity in comparison with imipenem. According to microbiological test data the (4S, 8S)-4-methoxy derivative showed the best activities. This research group extended their synthesis and studies to further 4-substituted derivatives of the GV104326 molecule. In these projects Andreotti D et al., [23], on the course of multistep procedure, the 2-(methoxyethoxy)-cyclohex-2-en-1-one (Figure 7, R = Me) or the epoxide derivative of the 4-cyclohexylazetidin-2- one was used as key compound for the construction of the cyclohexane ring (Figure 7, R = OH, F, CN). Compounds (Figure 7) have shown a good activity (MIC 0.03- 8 μg/ml) againts several bacterial strains.
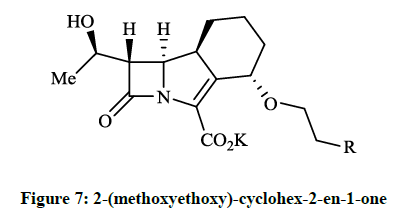
Figure 7: 2-(methoxyethoxy)-cyclohex-2-en-1-one
Fabio RD et al., [24] reported a careful preparation of 2- and 3-aminoethyloxy analogue of molecule GV104326. The 2-aminoethyloxy derivative was the most active compound (MIC 4 mg/ml) against Pseudomonas aeruginosa. Géhanne S et al., [25] achieved synthesis of 4- ureido trinems (Figure 8) from the known amino alcohol intermediate via Wittig-type cyclisation and followed by reaction with the corresponding isocyanate. The obtained 4-N-methyl-N’-alkyl ureido trinems had good antibacterial activity in the course of in vitro microbiological tests (MIC ≤ 0.12-8 μg/ml).
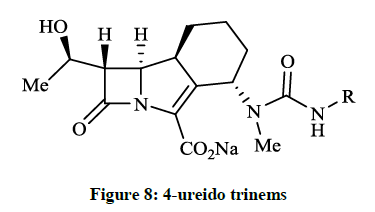
Figure 8: 4-ureido trinems
Kanno O et al., [26] described the synthesis and biological evaluation of the 4-pyrrolidin-3-ylthiomethyl derivative of GV104326 trinem (Figure 9). The authors established that compunds showed potent antimicrobial activity (MIC~1.5 μg/ml) against gram positive bacteria including S. aureus 535 (MRSA) and higher in vivo efficiency against S. aureus 507 (MIC 0.78 μg/ml).
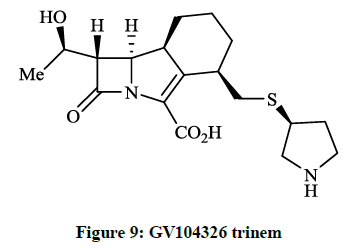
Figure 9: GV104326 trinem
When a bacteria produce β-lactamase enzymes, they hydrolyze the β-lactam ring of the antibiotic, thus may render a drug inactive. The 4- substituted trinems (tribactams) inhibit the broad spectrum β-lactamase enzymes and these compounds have different antibiotic activity generally. Copar A et al., [27] reported design, synthesis and pharmaceutical evaluation of novel unsubstituted trinems and their 6-thia derivatives (Figure 10, X = CH2 or S). Some products showed activity at the enzyme β-lactamase I from Bacillus cereus and another class A β- lactamase (E. coli TEM1). 38-61% Inhibition of β-lactam hydrolysis was observed in 100 μmol/l of lactamase enzyme I from B. cereus.
Plantan I et al., [28] described the structure-based design, synthesis and biological activity of several stereoisomeric 4(S or R)-alkoxy trinems (Figure 11), which are analogue of the lead compound (R = 4(S)-OMe, namely LK-157). The β-lactamase inhibitory activity of the derivatives (OEt, OPri, OBu, etc.) compared to LK-157 are decreased despite various lipophilicity. When Paukner S et al., [29] tested LK-157 againts the purified class A (TEM-1 and SHV-1) β-lactamases, ~50% inhibitory concentrations (IC50 ~55 and ~151 nM) were observed, furthermore the inhibition of the AmpC enzyme was significant on average (IC50 ~62 nM).
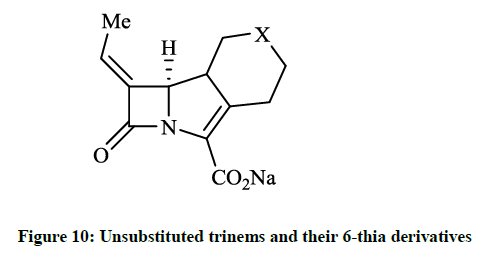
Figure 10: Unsubstituted trinems and their 6-thia derivatives
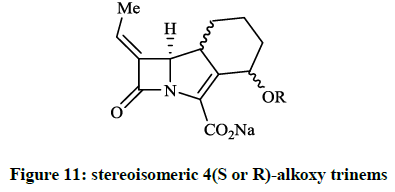
Figure 11: stereoisomeric 4(S or R)-alkoxy trinems
Isoindolinyl-4-oxoquinoline-3-carboxylic acids
Besides tricyclic β-lactams the quinolone-carboxylic acid derivatives represent important members of the large group of broad-spectrum active antibacterial drugs. These are active against both gram positive and gram negative bacteria and the compounds function by inhibiting DNA gyrase and the topoisomerase IV, which is one of two the type-II topoisomerases in bacteria. Todo Y et al., [30,31] patented an invention related to the synthesis of bioactive 7-isoindolinyl-quinolone-carboxylic acid derivatives, where a 6-nonfluorinated 1-cyclopropyl-8-(difluoromethoxy)- 7-(1-methylisoindolin-5-yl)-4-oxoquinoline-3-carboxylic acid (formally T-3811 and BMS-284756 or Garenoxacin) (Figure 12) was found as the candidate compound.
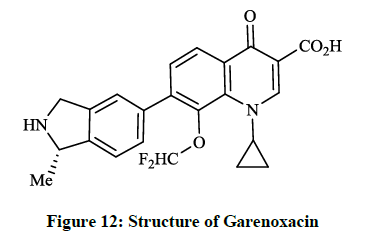
Figure 12: Structure of Garenoxacin
General synthetic route to 1-cyclopropyl-7-(isoindolin-5-yl)-4-oxoquinoline-3-carboxylic acids was achieved via Stille-type coupling reaction between N-Cbz-1-methyl-5-(tributylstannyl)isoindoline and ethyl 7-bromo-1-cyclopropyl-4-oxoquinoline-3-carboxylate was described by Reuman M et al., [32]. The alternative method was also reported by Todo Y et al., in patents [30,31] where the inventors applied the Suzuki cross-coupling reaction of the 7-[1-methyl-2-(triphenylmethyl)isoindolin-5-yl]boronic acid with ethyl 7-bromo-1-cyclopropyl-4-oxoquinoline-3- carboxylate. Takahata M et al., [33] investigated this so-called des-F(6)-quinoline compound by in vitro and in vivo methods for evaluation of antibacterial activity in comparison to other fluoroquinolones (ciprofloxacin, levofloxacin, trovafloxacin). T-3811 exhibited potent activity against S. aureus (MIC 0.025 μg/ml), the methicillin-resistant Staphylococcus epidermis (MIC 6.25 μg/ml), S. pneumoniae (MIC 0.05 μg/ml) and against M. tuberculosis was exceptionally effective (MIC 0.0625 μg/ml). In the course of in vitro studies of BMS-284756 (T-3811) Rhomberg PR et al., [34] observed high activity against Campylobacter jejuni, Helicobacter pylori, Legionella spp. (MIC50 0.032; 0.004 and 0.25 μg/ml) micro aerophillic and anaerobic bacteria. The results demonstrated that BMS-284756 had comparable or greater activity than other investigated quinolones. Hayashi K et al., [35] descibed the synthesis of a series of 1-cyclopropyl-7-(isoindolin-5-yl-4-oxoquinoline-3- carboxylic acids (Figure 13), their antibacterial and toxicological evaluation. The various substituents of the compounds prepared are R1, R2= H or/and CH3, R3= H, OCH3, OCHF2, while R4= H or F and the synthetic procedures were similar to the methods, that Todo Y et al., described [30,31].
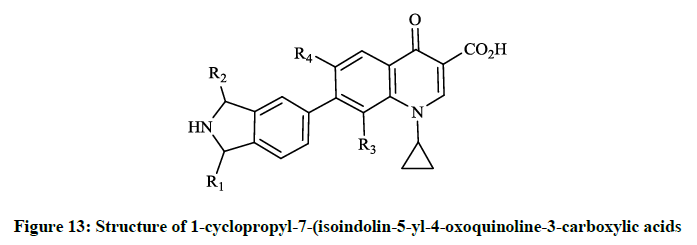
Figure 13: Structure of 1-cyclopropyl-7-(isoindolin-5-yl-4-oxoquinoline-3-carboxylic acids
Earlier Yatsunami T et al., [36] reported the synthesis and evaluation of antibiotic effects of several 7-(2-isoindolinyl)-1-cyclopropyl-6,8- difluoro-1,4-dihydro-4-oxoquinoline-3-carboxylic acid derivatives (Figure 14). Generally, the reaction of 6,7,8-trifluoro-dihydro-4- oxoquinoline-3-carboxylic acids with the corresponding isoindoline in DMF at 120°C for 1.5 h gave the desired products in good yields. This compound showed remarkable antibacterial activity against S. aureus, E. coli and P. aeruginosa (MIC <0.025 μg/ml; 0.2 μg/ml and 0.39 μg/ml). Similarly, Petersen U et al., [37] synthesized the tetra-hydro-isoindolinyl analogue (Figure 15), which according to the invention also exhibits surprising increases in action against the above-mentioned bacteria and Enterococcus faecalis.
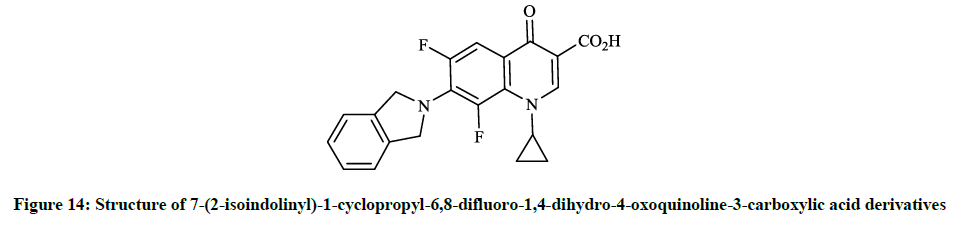
Figure 14: Structure of 7-(2-isoindolinyl)-1-cyclopropyl-6,8-difluoro-1,4-dihydro-4-oxoquinoline-3-carboxylic acid derivatives
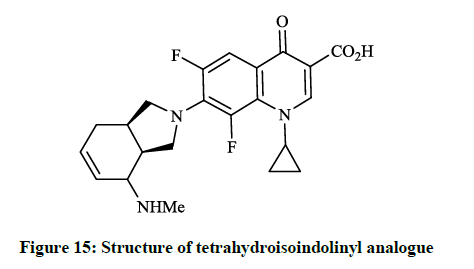
Figure 15: Structure of tetrahydroisoindolinyl analogue
Isoindolinone and its fused heterocycles
Mohamed EK et al., [38] synthesized some novel condensed pyrimidine derivatives using the 3-amino-1,9-dioxo-1,9-dihydropyrimido[6,1- a]isoindole-4-carbonitrile as a key intermediate. Thus tetracyclic pyrimido[6,1-a]isoindole-2-carboxylate (Figure 16) was prepared by the reaction of this amino-carbonitrile with diethyl malonate. The condensation of the key compound nitrile with triethyl orthoformate in refluxing acetic anhydride gave pyrimido[6,1-a]isoindolylimidoformate (Figure 17) and the reaction of aminocarbonitrile with 1,2-ethylenediamine in the presence of carbon disulfide afforded 4-(imidazol-2-yl)pyrimido[6,1-a]isoindole-1,9-dione (Figure 18).
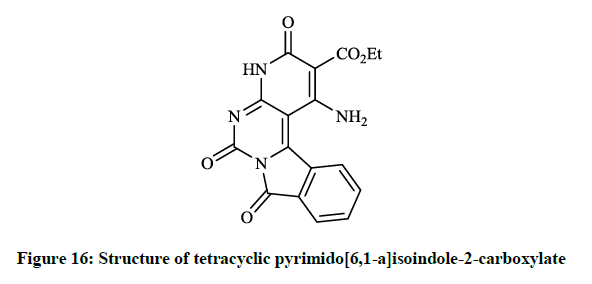
Figure 16: Structure of tetracyclic pyrimido[6,1-a]isoindole-2-carboxylate
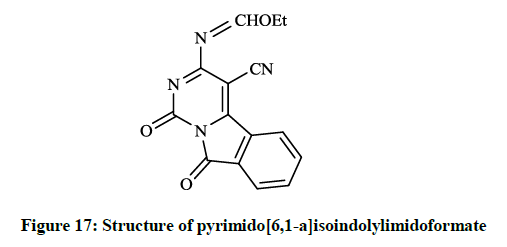
Figure 17: Structure of pyrimido[6,1-a]isoindolylimidoformate
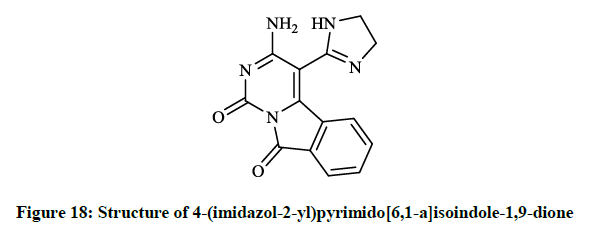
Figure 18: Structure of 4-(imidazol-2-yl)pyrimido[6,1-a]isoindole-1,9-dione
The preliminary bioactivity screening of these compounds was performed at 50 μg/ml against S. aureus and E. coli and results showed that imidazolyl derivative (Figure 18) and imidoformate derivative (Figure 17) had significant antimicrobial activities, while the tetracyclic isoindolone derivative (Figure 16) and the imidoformate derivative (Figure 17) had medium activity. Sui Z et al., [39] described synthesis of a series of isoindolo[2,1-a]quinolines from the reaction of substituted phthalic anhydrides with 2-aminoacetophenones in refluxing xylene. Demethylation of tetracyclic compounds was carried out with pyridine hydrochloride resulting in potent DNA-gyrase and topoisomerase-II inhibitor agents (Figure 19).
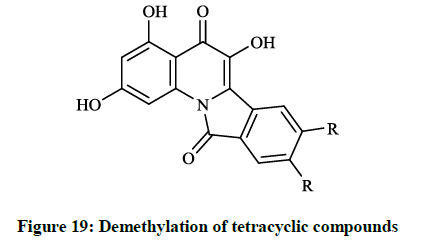
Figure 19: Demethylation of tetracyclic compounds
When substituents were changed to dichloro or dihydroxy (R = Cl, OH) on the isoindolone moiety, the compounds showed very good activity in bacterial DNA-gyrase assay (IC50 = 82 μg/ml and 22 μg/ml) and in human topoisomerase II assay (IC50 =13 μg/ml and 0.29 μg/ml), thus some of them are more potent than ellipticine or nalidixic acid. Lübbers T et al., [40] reported design and simple synthesis of isoindolo[2,1- a]quinolinones (Figure 20; R1 = Cl, R2 = OH or R1 = OMe, R2 = H) and isoindolinone fused 1,4-dihydro-2H-pyrido[3,2-d][1,3]oxazines (Figure 21; R1 = H, R2 = Me or R1 = 5,6-(CH)4, R2 = H ). Latter compounds (Figure 21) were proved to be effective DNA gyrase inhibitor in the supercoiling assay in vitro E. coli tests, the maximal non-effective concentration was low (MNEC = 0.13 μg/ml).
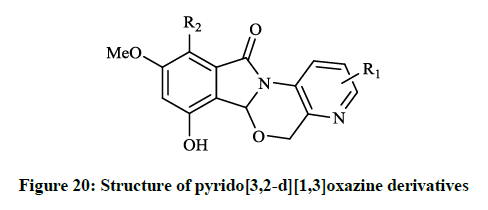
Figure 20: Structure of pyrido[3,2-d][1,3]oxazine derivatives
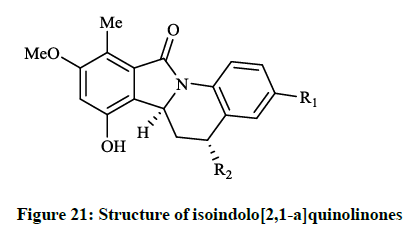
Figure 21: Structure of isoindolo[2,1-a]quinolinones
The in vitro antibiotic activities of pyrido[3,2-d][1,3]oxazine derivatives (Figure 20) were remarkable against S. aureus strains (MIC = 0.5-2.0 μg/ml), while isoindolo[2,1-a]quinolinones (Figure 21) had moderate activity against same bacteria. Cyclo-condensation reaction of methyl 2- formylbenzoate with various anilines under basic conditions and subsequent addition of the obtained aminal to thiols or electron rich heteroarenes, including furans, indoles resulted in novel 2,3-dihydroisoindolones. The allyl, 2-oxopropanyl, formylmethyl and hydroxyethyl derivatives were also prepared in further reactions, however these compounds showed moderate DNA gyrase inhibition or were almost inactive. These isoindolones led to the thorough SAR study of the novel phenolic DNA gyrase inhibitors. Narsimha S et al., [41] prepared a series of the imidazo[2,1-a] isoindolium bromide derivatives (Figure 22) from 2-iodobenzoic acid and N,N-carbonyldiimidazole (CDI) in a one-pot palladium-catalyzed coupling reaction in good yields.
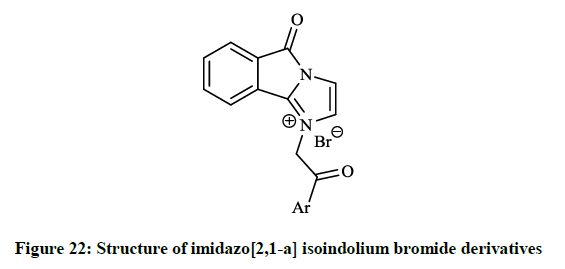
Figure 22: Structure of imidazo[2,1-a] isoindolium bromide derivatives
All the synthesized compounds were screened for their in vitro antibacterial activity and two derivatives showed excellent inhibition against both gram positive and gram negative bacteria (Table 1).
| Compound | Staphylococcus aureus | Bacillus subtilis | Escherchia coli | Proteus vulgaris |
|---|---|---|---|---|
| 1 (Ar = 4-Me-Ph-) | 3.125 | 25 | 12.5 | 6.25 |
| 2 (Ar = 4-Ph-Ph-) | 3.125 | 3.125 | 12.5 | 6.25 |
| 3 Streptomycin | 6.25 | 6.25 | 6.25 | 3.125 |
MIC: Minimum Inhibitory Concentration
Table 1: In vitro antibacterial activity (MIC in μg/ml) of 5H-imidazo[2,1-a]isoindolium bromides
Earlier Breytenbach JC et al., [42] reported the synthesis and in vitro antibacterial activity of a range of N-substituted isoindolin-5-ones (Figure 23; R= Me, Pri, Bui, Ph, etc). The tested compounds were prepared by refluxing α-amino acids (e. g. stereoisomers of alanine, valine, serine and aspartic acid) with o-phthalaldehyde under mild and simple reaction conditions.
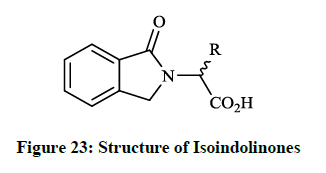
Figure 23: Structure of Isoindolinones
Isoindolinones (Figure 23) exhibited promising antibacterial activity (MIC 0.328-3.6 mg/ml) against B. subtilis, S. aureus, Micrococcus roseus and E. coli bacteria similar to β-lactam antibiotics. The carboxyl functional group in compounds seems to be necessary for antibacterial activity.
Conclusion
Over the past few decades numerous isoindolines and their fused derivatives were developed, synthesized and tested for antibacterial activities and the inhibition of pathogen bacteria reproduction. The mechanism of action of the synthesized and tested compounds was usually different, but sometimes the mechanism is complex or unknown so far. It may refer to inhibition of cell growth by interaction and modulation of a direct bio molecular target, such as a specific protein (enzyme, receptor) or nucleic acid (DNA). Hopefully, these results will serve support the medicinal chemists, researchers to design, coordinate and accomplish new approaches towards discovery of novel drugs.
Acknowledgement
The authors are would like to thank G. Csende for his careful revision of manuscript and his useful remarks and we also would like to express our thanks to the Institute of Pharmaceutical Chemistry, University of Szeged for the professional help.
References
- F. Csende, F. Miklós, G. Stájer, Curr. Org. Chem., 2012, 16(8), 1005, and references therein.
- R. Bonnet, M.B. Hursthouse, S.A. North, J. Trotter, J. Chem. Soc., Perkin Trans. 2, 1985, 293.
- R.K. Bhatia, Curr. Top.Med. Chem., 2017, 17(2), 189.
- T.S.A. Heugebaert, B.I. Roman, C.V. Stevens, Chem. Soc. Rev., 2012, 41, 5626.
- C. Yu, Q. Wu, J. Wang, Y. Wei, E. Hao, L. Jiao, J. Org. Chem., 2016, 81(9), 3761.
- Y. Ni, J. Wu, Org. Biomol. Chem., 2014, 12, 3774.
- H. Liu, Y. Wu, Z. Li, H. Lu, J. Porphyrins Phthalocyanines,2014, 18, 679.
- C.J.L. Murray, Lancet, 2016, 388, 1459.
- H. Neumann, D. Strübink, M. Lalk, S. Klaus, S. Hübner, A. Spannenberg, U. Lindequist, M. Beller, Org. Biomol. Chem., 2006, 4, 1365.
- A. Mikolasch, S. Hessel, M.G. Salazar, H. Neumann, K. Manda, D. Gördes, E. Schmidt, K. Thurow, E. Hammer, U. Lindequist, M. Beller, F. Schauer, Chem. Pharm. Bull., 2008, 56(6), 781.
- J.V. Heck, B. G. Christensen, Tetrahedron Lett., 1981, 22(50), 5027.
- B.G. Christensen, J.V. Heck, M. J. Szymonifka, US Patent 4, 374, 848, 1983.
- E. Di Modugno, I. Erbetti, L. Ferrari, G. Galassi, S.M. Hammond, L. Xerri, Antimicrob. Agents Chemother., 1994, 38(10), 2362.
- R. Wise, J.M. Andrews, N. Brenwald, Antimicrob. Agents Chemother., 1996, 40(5), 1248.
- K.V. Singh, T.M. Coque, B.E. Murray, Antimicrob. Agents Chemother., 1996, 40(9), 2142.
- S. Tamura, S. Miyazaki, K. Tateda, A. Ohno, Y. Ishii, T. Matsumoto, N. Furuya, K. Yamaguchi, Antimicrob. Agents Chemother., 1998, 42(7), 1858.
- B. Tamburini, A. Perboni, T. Rossi, D. Donati, D. Andreotti, G. Gaviraghi, R. Carlesso, C. Bismara, EP Patent 0416 953 (1991).
- M. Sendai, T. Miwa, EP Patent 0422 596 (1995).
- S. Michiyuki, T. Suita, US Patent 5, 393, 751 (1995).
- M. Sendai, T. Miwa, US Patent 5, 459, 260 (1995).
- I. Plantan, A. Prezelj, U. Urleb, B. Mohar, M. Stephan, EP Patent 2135 871 (2009).
- D. Andreotti, T. Rossi, G. Gaviraghi, D. Donati, C. Marchioro, E. Di Modugno, A. Perboni, Bioorg. Med. Chem. Lett., 1996, 6(4), 491.
- D. Andreotti, S. Biondi, R. Di Fabio, D. Donati, E. Piga, T. Rossi, Bioorg. Med. Chem. Lett., 1996, 6(16), 2019.
- R. Di Fabio, D. Andreotti, S. Biondi, G. Gaviraghi, T. Rossi, Bioorg. Med. Chem. Lett., 1996, 6(16), 2025.
- S. Géhanne, E. Piga, D. Andreotti, S. Biondi, D. Pizzi, Bioorg. Med. Chem. Lett., 1996, 6(22), 2791.
- O. Kanno, Y. Shimoji, S. Ohya, I. Kawamoto, J. Antibiot.,2000, 53(4), 404.
- A. Copar, T. Prevec, B Anžič, T. Mesar, L. Selič, M. Vilar, T. Solmajer, Bioorg. Med. Chem. Lett., 2002, 12(6), 971.
- I. Plantan, L. Selič, T. Mesar, P. Š. Anderluh, M. Oblak, A. Preželj, L. Hesse, M. Andrejašič, M. Vilar, D. Turk, A. Kocijan, T. Prevec, G. Vilfan, D. Kocjan, A. Čopar, U. Urleb, T. Solmajer, J. Med. Chem., 2007, 50(17), 4113.
- S. Paukner, L. Hesse, A. Preželj, T. Šolmajer, U. Urleb, Antimicrob. Agents Chemother., 2009, 53(2), 505.
- Y. Todo, K. Hayashi, M. Takahata, Y. Watanabe, H. Narita, WO Patent 029 102 (1997).
- Y. Todo, K. Hayashi, M. Takahata, Y. Watanabe, H. Narita, US Patent 6, 025, 370 (2000).
- M. Reuman, S.J. Daum, B. Singh, M.P. Wentland, R.B. Perni, P. Pennock, P.M. Carabateas, M.D. Gruett, M.T. Saindane, P.H. Dorff, S.A. Coughlin, D.M. Sedlock, J.B. Rake, G.Y. Lesher, J. Med. Chem., 1995, 38(14), 2531.
- M. Takahata, Y. Yamashiro, M. Yonezawa, H. Araki, Y. Todo, S. Minami, Y. Watanabe, H. Narita, Antimicrob. Agents Chemother., 1999, 43(5), 1077.
- P.R. Rhomberg, D.J. Biedenbach, R.N. Jones, Diagn. Microbiol. Infect. Dis., 2001, 40(1-2), 45.
- K. Hayashi, M. Takahata, Y. Kawamura, Y. Todo, Arzneim.- Forsch./Drug Res., 2002, 52(12), 903.
- T. Yatsunami, A. Yazaki, S. Inoue, H. Yamamoto, M. Yokomoto, Y. Nomiyama, S. Noda, EP Patent 0343 560 (1989).
- U. Petersen, A. Krebs, T. Schenke, F. Kunisch, T. Philipps, K. Grohe, K.D. Bremm, R. Endermann, K.-G. Metzger, I. Haller, H.J. Zeiler, US Patent, 1995, 464, 796
- E.K. Mohamed, W.S. Shehab, J. Korean Chem. Soc., 2011, 55(6), 988.
- Z. Sui, J. Altom, V.N. Nguyen, J. Fernandez, J.I. Bernstein, J.J. Hiliard, J.F. Barett, B.L. Podlogar, K. A. Ohemeng, Bioorg. Med. Chem., 1998, 6(6), 735.
- T. Lübbers, P. Angehrn, H. Gmünder, S. Herzig, Bioorg. Med. Chem. Lett., 2007, 17(16), 4708.
- S. Narsimha, K. Battula, N.V. Reddy, Synth. Commun., 2017, 47(9), 928.
- J.C. Breytenbach, S. van Dyk, I. van den Heever, S.M. Allin, C. C. Hodkinson, C.J. Northfield, M.I. Page, Bioorg. Med. Chem. Lett., 2000, 10(15), 1629.



