Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 6
2-[Benzyl(methyl)amino]propanedinitrile-New Building Block for Synthesis of Riociguat, a Pulmonary Hypertension Drug
Kristine Nerkararyan1, Karine Sargsyan1, Nairi Gasparyan1, Mariam Gharibyan1, Mikayel Movsisyan1* and Frank Porstmann2
1AZAD Pharmaceuticals LLC R&D Laboratory, Azatutyan ave. 26, Yerevan, Armenia
2AZAD Pharmaceutical Ingredients AG, Durachweg 15, CH-8200, Schaffhausen, Switzerland
- *Corresponding Author:
- Mikayel Movsisyan
AZAD Pharmaceuticals LLC R&D Laboratory
Azatutyan ave. 26, Yerevan, Armenia
Abstract
Riociguat is a potent, oral stimulator of soluble guanylate cyclase for the treatment of pulmonary hypertension, is prepared via an efficient, non-infringing synthesis route from commercially available diethyl malonate via [N-Methyl(benzyl)amino]propanedinitrile.
Keywords
Pulmonary hypertension (PH), Pulmonary arterial hypertension (PAH), Riociguat, Diethyl malonate, Diethyl bromomalonate, N-Methyl( benzyl)amine, Diethyl [N-methyl(benzyl)amino]malonate, [N-Methyl(benzyl)amino]-malonamide, [N-Methyl-(benzyl)amino]propane-dinitrile, 1-[(o-Fluoro-phenyl)-methyl]-1,2,7-triaza-1H-indene-3-carbo-xamidine.
Introduction
High blood pressure in the lungs is called Pulmonary Hypertension (PH) or Pulmonary Arterial Hypertension (PAH). It is characterized by increased blood pressure in pulmonary capillaries leading to reduced right heart function and eventual heart failure. Symptoms include shortness of breath, syncope, tiredness, chest pain, swelling of the legs, and a fast heartbeat. PAH is a chronic and life-changing disease that can lead to right heart failure if left untreated [1-3]. In all its variant presentations, this disease is estimated to affect up to 100 million people worldwide [4]. Endothelial dysfunction of the pulmonary vasculature plays a key role in the progression of PAH and is characterized by impaired production of vasodilators such as nitric oxide (NO) which is a key regulator of flow-induced vasodilation in the lung [5-8]. Treatment of PAH with Nitric oxide (NO)-releasing agents such as nitrates has failed to produce beneficial long-time effects [9]. There are three therapeutic target pathways in PAH: a) the prostacyclin pathway (e.g., prostanoids such as epoprostenol, iloprost, treprostinil); b) the endothelin pathway (eg, endothelin receptor antagonists [ERAs] such as bosentan, ambrisentan, macitentan); and c) the cyclic guanosine monophosphate (cGMP) degradation pathway (e.g., phosphodiesterase-5 [PDE5] inhibitors such as sildenafil and tadalafil) [10-13]. Despite the availability of numerous treatment options, the phosphodiesterase-5 inhibition is not effective in all the patients and mortality rates remain high [14-17].
Riociguat-1, a soluble guanylate cyclase (sGC) stimulator approved by the US Food and Drug Administration (FDA) in 2013 for the treatment of pulmonary arterial hypertension (PAH) and the treatment of chronic thromboembolic pulmonary hypertension [18]. Riociguat-1, offers a new mode of action since it has a dual mode of action that enhances sGC response to endogenous NO and directly stimulates sGC independently of NO binding [19]. It is the first member of novel class of therapeutics called sGC stimulator.
Several synthetic methods for 1 preparation were reported in the recent years, especially based on amidine intermediate 2, which was synthesized from 2-chloronicotinic acid 3 (Method A) [20], 2-fluoro-3-(trifluoroacetyl)pyridine 4 (Method B) [21] and ethyl cyanopyruvate sodium salt 5 (Method C) [22]. C. Hirth-Dietrich and co-workers proposed an alternative approach, avoiding usage of the intermediate 2, based on coupling of pyrazolopyridine derivative 6 and 2-Chloro-5-nitro-4,6-pyrimidinediamine 7 in the presence of hexabutylditin and catalytical amount of Tetrakis (triphenylphosphine) palladium (Method D) (Scheme 1) [23]. However, the most convenient and straightforward synthetic approach to 1, involves coupling of amidine intermediate 2 with a 2-substituted malononitrile building block. Phenylazomalononitrile 8 is used as a coupling pair in all known synthetic approaches based on amidine intermediate 2, leading to the corresponding coupling product–pyrimidine derivative 9. Despite the advantages of the mentioned approach, reduction of the pyrimidine derivative 9 to the corresponding 4,5,6- triaminopyrimidine derivative 10, requires harsh conditions, especially, up to 65 bar of hydrogen pressure.
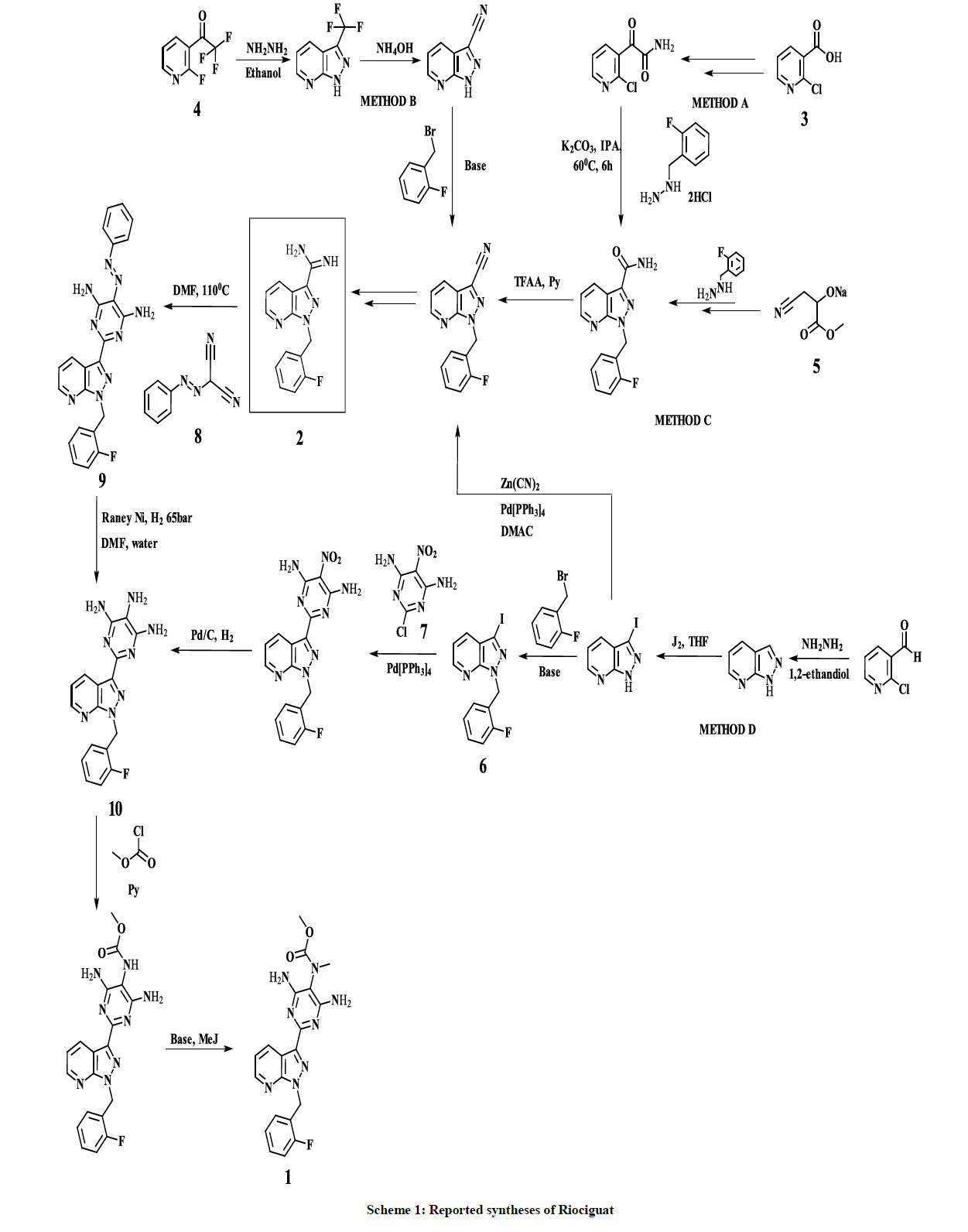
Scheme 1: Reported syntheses of Riociguat
In this article, we report synthesis of [N-Methyl(benzyl)amino]propanedinitrile 11, which is used as a 2-substituted malononitrile building block in the coupling step with the amidine intermediate 2 for the preparation of Riociguat in much milder conditions and beneficial conditions for Active Pharmaceutical Ingredient (API) synthesis i.e. avoidance of highly toxic reagent or metal catalysts.
Materials and Methods
All reagents and solvents were obtained commercially from abcr GmbH and were used without further purification. Diethyl bromomalonate 12 is synthesized according to known procedure, via bromination of diethylmalonate 13 [24]. 1-(2-Fluorobenzyl)-1H-pyrazolo[3.4-b]pyridine-3- carboximidamide 2 is synthesized according to known procedure [25] and isolated as hydrochloride salt. Melting points were determined on a Buchi B-545 melting point apparatus in open capillaries. Thin Layer Chromatography (TLC) analysis is performed using Silicagel 60 F254 Merck plates, Phenomenex Luna C18 (2), 150 x 3 mm, 5 μm column was used for High Performance Liquid Chromatography (HPLC) analysis. 1H-NMR (300 MHz) and 13C-NMR (75 MHz) spectra were recorded in d6-DMSO using a Varian Mercury-300VX instrument and TMS as an internal standard.
Diethyl [N-methyl(benzyl)amino]malonate HCl salt (14)
To a solution of diethyl bromomalonate 12 (22.18 g, 0.09278 mol) in 50 ml of ethanol N-methyl(benzyl)amine 15 (22.46 g, 0.1853 mol) was added drop wise while stirring in the course of 15 min. The yellow reaction mixture was heated under reflux for one hour. The solvent was evaporated in vacuum and 50 ml of diethyl ether were added to the resulting yellow slurry, triturated and allowed to stay at RT for 2 h. The precipitated solid (N-methyl(benzyl)amine hydrobromide) was filtered off with suction and the filter cake was washed with diethyl ether three times. To the combined ethereal washings 32 ml of HCl saturated ethereal solution were added drop wise in the course of 15 min while stirring and the resulting suspension was allowed to stay at ambient temperature overnight. The precipitated white solid was filtered off with suction, washed four times with diethyl ether and dried in vacuum to give the title product as white powder (24.35 g, yield 83%, 90% HPLC peak area); 1H-NMR (300 MHz, d6-DMSO): 1.31 (6H, t, J=7.1 CH3), 2.59 (3H, s, NCH3), 4.08 (2H, s, NCH2), 4.24 (4H, q, J=7.1, CH2), 4.49 (1H, C, NCH), 7.24-7.35 (3H, m, Ph), 7.44-7.49 (2H, m, Ph), 8.68 (2H, br, HCl).
[N-Methyl(benzyl)amino]malonamide (16)
100 ml sodium bicarbonate 10% aqueous solution was added to Diethyl [N-methyl(benzyl) amino]malonate HCl salt (14) (15.75 g, 0.04987 mol) and stirred vigorously for 5 min. The phases were separated and the aqueous phase was extracted with diethyl ether. Combined organic washings were dried over MgSO4, filtered and evaporated to dryness. To the residual 12.6 g of milky-white oil, formamide (11.675 g) and DMF (60.5 ml) were added and the mixture was heated to 100°C. A suspension of sodium methoxide (3.12 g, 0.05775 mol) in 50 ml of methanol was added to the reaction mixture in the course of 20 min and heated under reflux for 2.5 h. The reaction mixture was evaporated to dryness in vacuum, to the residue 100 ml acetone/diethyl ether 1:1 mixture was added and stirred for 30 min. The precipitated solid was filtered off with suction, the filter cake was washed with ether three times (25 ml each). The filtrate was evaporated to approx. 1/8, 10 ml of acetonitrile were added and solution was kept in a fridge for 24 h. The precipitated solid was filtered off with suction and dried in vacuum resulting in the title product as white solid (4.42 g, yield 40%, 97.7% HPLC peak area); 1H-NMR (300 MHz, d6-DMSO): 2.23 (3H, s, CH3), 3.53 (1H, s, CH), 3.69 (2H, s, CH2), 7.11 (2H, br, NH2), 7.16-7.31(3H, m, Ph), 7.34-7.39(4H, m, Ph and NH2).
2-[Benzyl(methyl)amino]propanedinitrile (11)
2-[Benzyl(methyl)amino]propanediamide (16) (3.7 g, 0.0167 mol) was suspended in 80 ml THF and pyridine (6.2 g, 0.07838 mol) was added drop wise in the course of 10 min while stirring. Subsequently, TFAA (7.6 g, 0.03618 mol) was added portion wise and the reaction mixture state changed to a yellow-orange solution. The reaction mixture was stirred after completion of addition at ambient temperature for additional 10 min while total consumption of the starting material was confirmed by TLC (Dioxane/n-heptane 1: 1 Rf = 0.7). The reaction mixture was poured into 120 ml H2O and extracted four times with EtOAc (40 ml each). The combined organic washings were dried over MgSO4, filtered and concentrated in vacuum to give the crude product as light-yellow oil. Purification by column chromatography (mobile phase dioxane/heptane 1: 1) afforded the title product as colorless oil (2.52 g, yield 76.6%, 94% HPLC peak area); 1H-NMR (300 MHz, d6-DMSO): 2.37 (3H, s, CH3), 3.68 (2H, s, CH2), 5.71 (1H, s, CH), 7.25-7.37(5H, m, Ph); 13C-NMR (75.5 MHz, d6-DMSO): 37.9 (CH3), 47.1 (NCH), 58.2 (CH2), 110.4 (CN), 127.4 (CH), 128 (CH), 128.4(CH), 135.5 (CN).
N5-Benzyl-2-[1-[(2-fluorophenyl)methyl]pyrazolo[3,4-b]pyridin-3-yl]-N5-methyl-pyrimidine-4,5,6-triamine (17)
1-(2-Fluorobenzyl)-1H-pyrazolo[3.4-b]pyridine-3-carboximidamide (2) HCl salt (1.5 g, 0.0049 mol) was dissolved in 120 ml H2O, sodium carbonate (0.68 g, 0.00638 mol) was added and the mixture was stirred for 10 min. The reaction mixture was extracted three times with EtOAc, organic washings were dried over MgSO4, filtered and evaporated to dryness. The residue was taken up in 20 ml n-butanol followed by addition of pyridine (0.44 g, 0.00557 mol) and 2-Benzyl(methyl)amino]propanedinitrile (11) (1.03 g, 0.00557 mol). The reaction mixture was heated under reflux for 2.5 h when TLC (Dioxane/Heptane 1: 1 Rf = 0.47) confirmed total consumption of the starting materials. The reaction mixture was cooled to ambient temperature and evaporated to dryness. The resulting brownish viscous residue was triturated with diethyl ether, the precipitated solid was filtered off with suction and dried in vacuum to give the title product (1.58 g, yield 73%, 97% HPLC peak area, M. p. 190- 192°C); 1H-NMR (300 MHz, d6-DMSO): 2.63 (3H, s, NMe); 4.1 (2H, s, CH2, Bn); 5.78 (2H, s, CH2, F-Bn); 5.95 (4H, br, NH2); 7.02-7.4 (m, 10H (1,2,8-13)); 8.50 (1H, dd, (14)); 9.01-9.06 (1h, dd, (3)).
Riociguat (1)
A mixture of N5-Benzyl-2-[1-[(2-fluorophenyl)methyl]pyrazolo[3,4-b]pyridin-3-yl]-N5-methyl-pyrimidine-4,5,6-triamine (17) (1.5 g, 0.00334 mol), Pd/C 10% (3 g), ammonium formate (2.1 g, 0.0334 mol) and methanol (50 ml) was charged to an autoclave and stirred under 3.5 bar hydrogen pressure for 9 h at ambient temperature. The catalyst was filtered off and the filtrate was concentrated to 1/5 of the initial volume under reduced pressure. The precipitated white solid was washed with water, dried in vacuum and suspended in 15 ml 2-propanol. Dimethyldicarbonate (DMDC) (0.41 g, 0.00306 mol) was added and the suspension was stirred 24 h at ambient temperature. The reaction mixture was filtered with suction and the filter cake was dried in vacuum to give the crude product (0.76 g, 92.44% HPLC purity). Carbon treatment and recrystallization from MeOH afforded the title product (0.56 g, 97% HPLC purity, M. p. 257-259°C); 1H-NMR (300 MHz, d6- DMSO): 3.07 (3H, s, NCH3); 3.60 (2H, s) and 3.71 (1H, s, OCH3); 5.79 (2H, s, NCH2); 6.40 (4H, br, NH2); 7.11 (3H, m, C6H4); 7.24 (1H, dd, J = 8.0, 4.5, (2)); 7.30 (1H, m, C6H4); 8.60 (1H, dd, CH, J = 4.5, 1.6 (3)); 9.05-9.10 (1H, dd, J= 8.0, 1.5 (1)); 13C-NMR (75.5 MHz, d6-DMSO): 33.8 (NCH3); 43.2 (d, JC,F = 3.8, NCH2); 52.1 (OCH3); 99.4; 114.6 (CH, Py); 114.7 (d, JC,F = 21.1, CH, C6H4); 117.0 (CH, Py); 123.8 (d, JC,F = 3.5, CH, C6H4); 124.0 (d, JC,F = 14.4, CH, C6H4); 128.8 (d, JC,F = 8.0, CH, C6H4); 129.2 (d, JC,F = 3.9, CH, C6H4); 133.6 (2CNH2); 141.8; 147.9 (CH, Py); 150.8; 155.3; 157.0; 159.1; 159.6 (d, JC,F = 246.6, CF).
Results and Discussion
Despite the fact that compound 11 is potentially a very useful building block, only very few methods are known for its syntheses. S. Xiaobo and H. Yue described several synthetic approaches based on 2-bromomalononitrile 18 [26] (Scheme 2).
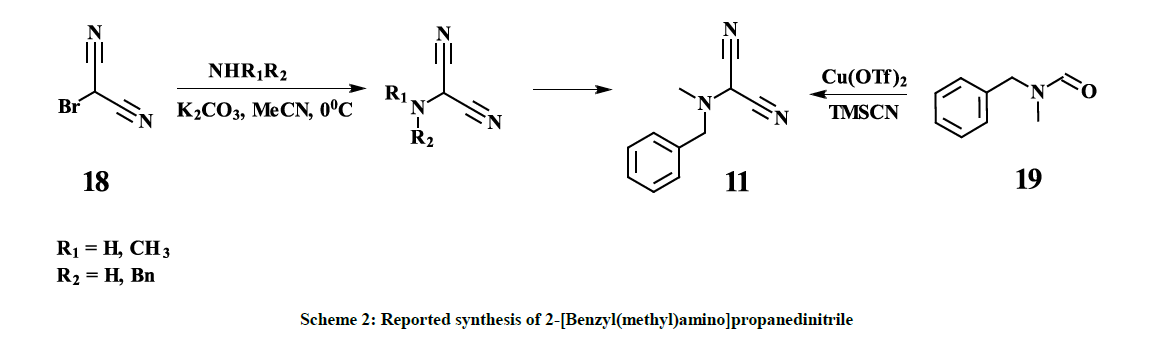
Scheme 2: Reported synthesis of 2-[Benzyl(methyl)amino]propanedinitrile
However, all our attempts to synthesize 11 following to the procedures described in [26] failed – only corresponding amine hydrobromides with almost quantitative yields along with some resin materials were isolated after interaction of 2-bromomalononitrile 18 [27] and the amine, which indicates high acidity of the hydrogen atom in 2-bromomalononitrile 18. Thus, so far, the only one known synthetic approach, reported by Xue- Qing Mou and co-authors [28], based on copper-catalyzed dicyanation of N-methyl-N-benzylformamide 19, provides 11 with 39% yield (Scheme 2). Despite the fact that the mentioned approach is a one-pot synthesis and seems to be straightforward, there are some obvious disadvantages, especially, commercial inaccessibility of the starting material 19, usage of expensive copper triflate and toxic trimethylsilanecarbonitrile. Furthermore, the removal of copper residues down to acceptable levels might be challenging. On the other hand, the above-mentioned disadvantages are not compensated by a high yield. Hereby, we report a four-step synthesis of 11 in mild conditions with overall 25% yield, based on commercially available and cheap starting materials – diethylmalonate 13 and N-methyl(benzyl)amine 15. Purification of the interim compounds is not complicated and is feasible in production. Obtained by this procedure 11 is successfully used as key intermediate in the three-step synthesis of Riociguat (1) with overall 40% yield. Diethyl-2-bromomalonate 12 [24] is converted to Diethyl [N-methyl( benzyl)amino]malonate HCl salt 14 with 83% yield. The Diethyl [N-methyl(benzyl)amino]malonate HCl salt 14 is converted to [N-Methyl( benzyl)amino]malonamide 16 with 40% yield and subsequently to 2-[Benzyl(methyl)amino]propanedinitrile 11 with 76.6% yield according to scheme 3.
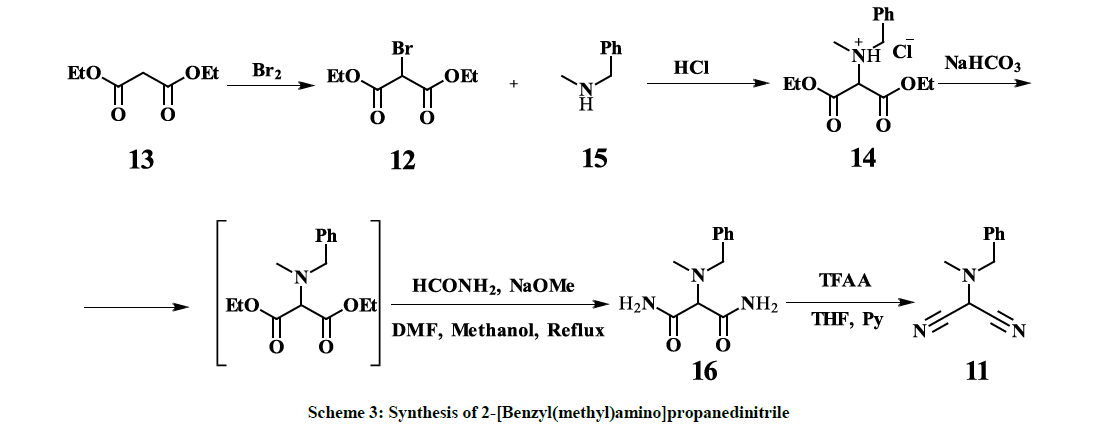
Scheme 3: Synthesis of 2-[Benzyl(methyl)amino]propanedinitrile
2-[Benzyl(methyl)amino]propanedinitrile 11 is reacted with the amidine key intermediate 2 to give N5-Benzyl-2-[1-[(2- fluorophenyl)methyl]pyrazolo[3,4-b]pyridin-3-yl]-N5-methyl-pyrimidine-4,5,6-triamine 17 with 73% yield. Catalytic reduction of the derivative 9, obtained by a similar coupling reaction of 2 with phenylazomalononitrile 8, to the corresponding 4,5,6-triaminopyrimidine derivative 10 requires up to 65 bar hydrogen pressure [22,25], meanwhile, subsequent catalytic benzyl-deprotection of 17 undergoes smoothly, already at 3.5 bar of hydrogen pressure. The Introduction of Moc-group is successfully performed by interaction of the interim benzyl-deprotected compound with DMDC, instead of highly toxic methyl chloroformate – which is used for the same purpose by C. Alonso-Alija and co-authors [22,25] – afforded finally Riociguat 1 with overall 40% yield as shown in scheme 4.
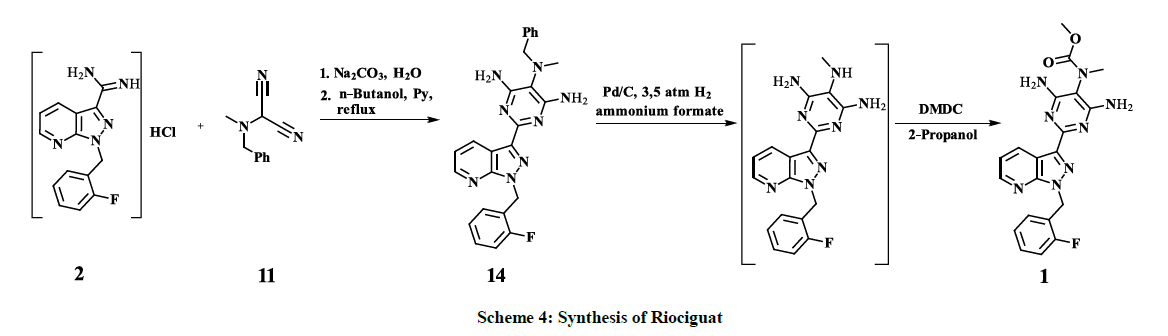
Scheme 4: Synthesis of Riociguat
Conclusion
2-[Benzyl(methyl)amino]propanedinitrile was synthesized in mild conditions with a yield comparable to the known methods, from commercially available, cheap starting materials and successfully used for Riociguat synthesis, avoiding harsh conditions, the usage of toxic reagents and homogenous metal catalysts. It can be also used as a convenient coupling pair with variety of amidine compounds and serve as a building block for synthesis of 2-substituted 4,6-diamnino-5-(methyl)aminopyrimidines.
Acknowledgement
The authors are grateful to AZAD Pharmaceuticals R&D management for allowing them to publish these results.
References
- S.P. Gaine, L.J. Rubin, Lancet., 1998, 352, 719-725.
- G. Simonneau, N. Galie, L.J. Rubin, J. Am. Coll. Cardiol., 2004; 43: 5S-12S.
- R.T. Schermuly, Nat. Rev. Cardiol., 2011, 8, 443-455.
- G. Simonneau, J. Am. Coll. Cardiol.,2009, 54, S43-S54.
- V.V. McLaughlin, M.D. McGoon, Circulation., 2006, 114, 1417-1431.
- J.P. Stasch, O.V. Evgenov, Handb. Exp. Pharmacol., 2013, 218279-313.
- H.H. Schmidt, U. Walter, Cell., 1994, 78(6), 919-925.
- J.R. Klinger, S.H. Abman, M.T. Gladwin, Am. J. Respir. Crit. Care Med., 2013, 188(6), 639-646.
- M. Andrew, A. Ian, L.W. David, Ann Thoracic Surg., 1996, 62, 1759-1764.
- N. Galie, M. Humbert, J.L. Vachiery, Eur. Heart J., 2016, 37(1), 67-119.
- N. Galiè M. Humbert.; J.L. Vachiery, Eur. Respir. J., 2015, 46(4), 903-975.
- M. Humbert, H.A. Ghofrani, Thorax., 2016, 71(1), 73-83.
- H.A. Ghofrani, R.Wiedemann, H. Olschewski, W. Seeger, F. Grimminger, Ann. Intern. Med.,2002, 136, 515-522.
- R.L. Benza, D.P. Miller, R.J. Barst, Chest., 2012, 142(2), 448-456.
- M. Humbert, O. Sitbon, A. Chaouat, Circulation., 2010, 122(2), 156-163.
- M. Humbert, O. Sitbon, A. Yaici, Eur. Respir. J., 2010, 36(3), 549-555.
- H.W. Farber, D.P. Miller, A.D. Poms, Chest., 2015, 148(4), 1043-1054.
- FDA Approves Riociguat for PAH and CTEPH. Medscape., 2013.
- H.A. Ghofrani, M. Humbert, D. Langleben, R. Schermuly, J.P. Stasch, M.R. Wilkins, J.R. Klinger, Chest., 2017, 151(2), 468-480.
- V. Arava, S. Gogireddy, Der Pharma Chemica.,2013, 5(4), 232-239.
- G. Depei, W. Qihua, CN 104086545, 2014.
- C. Alonso-Alija, U.S. Patent 7173037, 2007.
- C. Hirth-Dietrich, P. Sandner, J.P. Stasch, A. Knorr, G. Von Degenfeld, M. Hahn, M. Follmann, U.S. Patent 2014/0038956 A1, 2014.
- C.S. Palmer, P.W. McWherter, R. Adams, M.M. Brubaker, Organic Syntheses., 1941, 1, 245.
- J. Mittendorf, S. Weigand, C. Alonso-Alija, E. Bischoff, A. Feurer, M. Gerisch, A. Kern, A. Knorr, D. Lang, K. Muenter, M. Radtke, H. Schirok, K. Schlemmer, E. Stahl, A. Straub, F. Wunder, J.P. Stasch, Chem. Med. Chem., 2009, 4, 853-865.
- S. Xiaobo, H. Yue, CN 105367568, 2016.
- H. Takeshita, A. Mori, K. Kubo, Y. Qiu, A.B. Smith, Organic Syntheses., 1998, 75, 210.
- X.Q. Mou, L. Xu, S.H. Wang, C. Yang, Tetrahedron Letters., 2015, 56, 2820-2822.



